Commentary
Epidermal Growth Factor Receptor-Activating Mutation and Anaplastic lymphoma Kinase Gene Rearrangement as Oncogenic Co-Addiction: A Case Report of Adenocarcinoma Treated with Sequential Tyrosine Kinase-EGFR and ALK-Tyrosine Kinase Inhibitors
Benoit Roch1, vanessa Szablewski2, Isabelle Serre2, Julie Vendrell2, Jerome Solassol3 and Jean-louis Pujol1,3*
1Department of Thoracic Oncology, Arnaud de Villeneuve Hospital, Montpellier, France
2Department of Pathology, Arnaud de Villeneuve Hospital, Montpellier, France
3Cancer Research Institute of Montpellier, Montpellier, France
*Corresponding author: Jean-louis Pujol, Department of Thoracic Oncology, Montpellier Academic Hospital, Montpellier, France
Published: 08 Jul, 2018
Cite this article as: Roch B, Szablewski V, Serre I, Vendrell
J, Solassol J, Pujol J-l. Epidermal
Growth Factor Receptor-Activating
Mutation and Anaplastic lymphoma
Kinase Gene Rearrangement as
Oncogenic Co-Addiction: A Case
Report of Adenocarcinoma Treated with
Sequential Tyrosine Kinase-EGFR and
ALK-Tyrosine Kinase Inhibitors. Clin
Oncol. 2018; 3: 1482.
Abstract
Epidermal Growth Factor Receptor (EGFR)-activating mutation and Anaplastic Lymphoma Kinase (ALK) gene rearrangement are the two most frequently observed druggable oncogenic addictions in adenocarcinoma of the lung. Hitherto, they are considered as mutually exclusive and searching for ALK rearrangement when EGFR sensitizing mutation has been demonstrated is considered by many guidelines as not mandatory [1]. Recently coaltered EGFR and ALK genes in the same tumor have been described using micro-dissection techniques in two out of 629 adenocarcinomas [2]; this phenomenon is supposed to be linked with tumor heterogeneity. However, whether both altered genes behave as oncogenic drivers is not known. We report here a case of adenocarcinoma with concomitant EGFR/ALK gene alterations for which sequential therapy with EGFR-Tyrosine Kinase Inhibitors (TKI) and ALK-tyrosine kinase inhibitors demonstrated druggable status of both altered genes.
Case Presentation
In September 2015, a 68-year-old non-smoker Caucasian woman was diagnosed with a stage IV M1b lung cancer; bone and liver metastases were detected using computed assisted Positron Emission Tomography (PET/CT). Adenocarcinoma with massive features was demonstrated on a right lower lobe tumor transparietal biopsy. Genetic profiling using next-generation sequencing demonstrated an EGFR exon 21 mutation Leu858Arg with a variant allele frequency of 35% that was further confirmed by Sanger sequencing (Figure 1). On the same tumor specimen, an ALK over expression was detected by immunostaining using 5A4 clone and the presence of an ALK rearrangement was further confirmed by break-apart fluorescence in situ hybridization (Figure 2). Gefitinib was given as front line therapy obtaining clinical alleviation and dramatic tumor response. During the 9 following months, the patient benefited from a progression free interval. In May 2016, two brain metastases were detected as unique sites of progression and stereotaxic radiotherapy was performed. Liquid biopsy failed to show EGFR exon 20 Thr790Met mutation but still detected exon 21 Leu858Arg mutation. According to current guidelines, EGFR-TKI was maintained. The patient benefited from 5 additional progression free months. In September 2016, she experienced bone pains and fatigue and PET/CT demonstrated a systemic progressive disease: cervical, abdominal and mediastinal lymph nodes were observed together with new bone metastases whereas right lower lobe lung tumor and liver metastases were still controlled. Liquid biopsy and new transparietal biopsy did not detect EGFR exon 20 Thr790Met. Taking into account the initially known ALK translocation, the patient received Crizotinib 250 mg twice daily. She recovered a good clinical status and a new PET/CT showed evidence of response. Unfortunately, in December 2016, after only 4-months without progression, she experienced rapid growth of innumerous brain metastases and she then received Whole Brain Radiotherapy (WBRT). Lymph nodes and bones metastases that have motivated switch from EGFR-TKI towards ALK-TKI remained controlled. A third search for Thr790Met was positive. Crizotinib was stopped and Osimertinib therapy was started in January 2017. Unfortunately, the brain metastases were out of control despite WBRT and she died in March 2017.
Figure 1
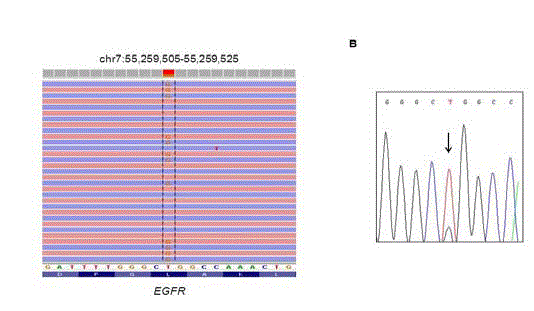
Figure 1
Identification of the EGFR exon 21 mutation Leu858Arg by nextgeneration
sequencing, visualized with the Integrative Genomic Viewer (1A)
and confirmed by Sanger sequencing (1B).The number of reads for the
reference allele and the variant allele, as well as the variant allele frequency
are displayed.
Figure 2
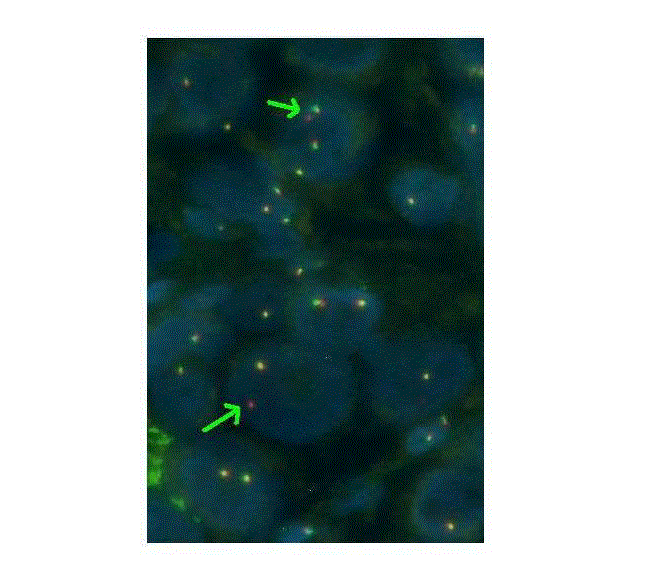
Figure 2
ALK FISH break-apart probe (Vysis) showing one normal fused
red/green signal and one isolated 3’ALK (single red signal - green arrow),
magnification x40.
Discussion
It is well-known that genotypic instability and the expression of cellular diversification mechanisms ensure that malignant neoplasms contain heterogeneous, phenotypically diverse tumor-cell subpopulations [3]. Apposition of different histological subtypes in the same primary lung neoplasm has been demonstrated since the 80’s but co-alteration of different oncogenic pathways is a more recent observation. Schmid et al. reported five patients with ALK/ EGFR coalteration. Outcomes with ALK and EGFR TKI seem inferior to what would be expected in patients with either alteration alone. However, the characterization of the ALK rearrangement was not optimal [4]. Reported a study in 629 lung adenocarcinoma primary tumors Cai et al. [2]: two of them presented with EGFR and ALK co-altered status. Interestingly, in these two cases, microdissection was performed in order to capture spatially separated tumor cell subpopulations according to adenocarcinoma subtypes. These different areas of the same recut section showed that ALK fusions did not coexist with EGFR mutations in all tumor cells with some areas presenting with both mutations whereas some others only had the EGFR mutation. One can argue that evidence of EGFR and ALK co-alteration as showed in the study by Cai et al. [2] or by Lee et al. [5], does not demonstrated dual EGFR/ALK oncogenic addiction. Chiari et al. [6] reported a case of a never smoker woman diagnosed with a stage IV lung adenocarcinoma who achieved long-term response to Gefitinib and several years later to Crizotinib. Gefitinib was active during 24 months with unknown mutational status at that time. EGFR exon 21 mutation Leu858Arg was detected two years later and ALK translocation was detected after one more year. ALK translocation was not detected on the tumors samples at diagnosis or at first recurrence. The disease was sensitive to Crizotinib. One can hypothesize that EGFR and ALK alterations have been temporarily separated rather than being dual oncogenic drivers at time of diagnosis inasmuch as resistance mechanisms to EGFR TKI can be lined by a bypass signaling pathway mediated by ALK and conversely [7]. In our case report, co-alteration was present in the primary tumor at time of diagnosis and both mutations were druggable. However, when dual oncogenic driver is observed, the standard therapy regimens may not be the optimal choice: the first-line then second-line concept of tumor therapy is at least questionable for our patient. As long as tolerance profile of EGFR TKI – ALK TKI combination is unknown, specific treatment of dual EGFR – ALK alterations remains challenging.
Figure 3
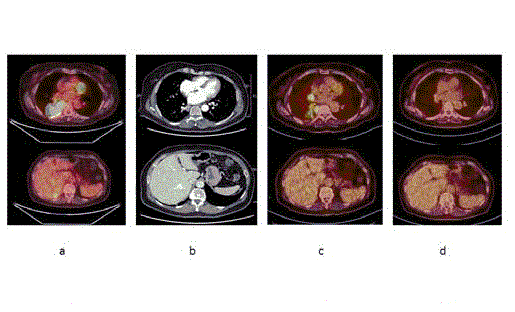
Figure 3
(3a) PET-scan at diagnosis. (3b) CT-scan demonstrating response
to Erlotinib. (3c) PET-scan at first progression. (3d) PET-scan demonstrating
response to Crizotinib.
References
- Jurgens J, Engel-Riedel W, Stoelben E, Schildgen V, Schildgen O, Brockmann M. The (Con-) Fusion in ALK Diagnostics: When Food and Drug Administration-Approved Algorithms Fail. J Clin Oncol. 2016;34(16):1961-2
- Cai W, Lin D, Wu C, Li X, Zhao C, Zheng L, et al. Hirsch FR. Intratumoral Heterogeneity of ALK-Rearranged and ALK/EGFR Coaltered Lung Adenocarcinoma. J Clin Oncol. 2015;33(32):3701-9.
- Nicolson GL. Tumor cell instability, diversification, and progression to the metastatic phenotype: from oncogene to oncofetal expression. Cancer Res. 1987;47(6):1473-87.
- Schmid S, Gautschi O, Rothschild S, Mark M, Froesch P, Klingbiel D, et al. Clinical outcome of ALK-positive non small cell lung cancer patients with de novo EGFR or KRAS co-mutations receiving tyrosine kinase inhibitors (TKI). J Thorac Oncol. 2017;12(4):681-688.
- Lee JK, Kim TM, Koh Y, Lee SH, Kim DW, Jeon YK, et al. Differential sensitivities to tyrosine kinase inhibitors in NSCLC harboring EGFR mutation and ALK translocation. Lung Cancer. 2012;77(2):460-3.
- Chiari R, Duranti S, Ludovini V, Bellezza G, Pireddu A, Minotti V, et al. Long-term response to gefitinib and crizotinib in lung adenocarcinoma harboring both epidermal growth factor receptor mutation and EML4- ALK fusion gene. J Clin Oncol. 2014;32(9):e30-2.
- Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71(18):6051-60.