Case Presentation
Extra Nodal RosaiDorfman Disease Mimicking Para Spinal Neoplasm - A Diagnostic Dilemma
Gautam Mukhopadhyay1, Suvadip Chakrabarti1* and Shilpi Kundu2
1Departmant of Surgical Oncology, Ruby Cancer Center, India
2Department of Pathology, Ruby General Hospital, India
*Corresponding author: Suvadip Chakrabarti, Departmant of Surgical Oncology, Ruby Cancer Center, India
Published: 20 Mar, 2018
Cite this article as: Mukhopadhyay G, Chakrabarti S,
Kundu S. Extra Nodal RosaiDorfman
Disease Mimicking Para Spinal
Neoplasm - A Diagnostic Dilemma. Clin
Oncol. 2018; 3: 1438.
Abstract
RosaiDorfman disease is a self-limiting rare histiocytic proliferative disorder of unknown disorder.
It classically presents with painless bilateral cervical lymphadenopathy. Extra nodal presentation
has been documented in the head neck region. We present a rare case RosaiDorfaman disease in
20 years old woman who presented with a painless para spinal mass for close to a year. This case is
being reported for is rarity and unusual anatomic presentation, which to our best knowledge has
not been reported in literature.
Keywords: RosaiDorfman disease; Para spinal mass; Histiocytic proliferative disorder
Introduction
Rosai-Dorfman Disease (RDD) is a rare and benign histiocytic proliferative disorder of unknown aetiology. It usually presents with painless lymphadenopathy with or without extra nodal manifestations.
Case Presentation
A 20 years old lady with history of a gradually progressive swelling over the right lower back for over a year. She had no history of similar swelling elsewhere over her body or any history of trauma over the back. No history similar complaints in first degree relatives. On examination there was a solitary, smooth, ovoid swelling 4 X 3 cm, mobile, non-fluctuant, painless, no impulse on coughing over right lower back in the para spinal region. Examination of local and regional lymph node was negative. CT scan of the Lower spine: Revealed a solitary 5.8 X 5.78 X 2.78 cm solitary hyperdense and enhancing lesion in the subcutaneous plane in the right posterior para spinal region without any extension or bony erosion or calcification or necrosis (Figure 1). FNAC of the swelling was suggestive of chronic inflammatory RosaiDorfman disease (Figure 2). Preoperative investigations were performed and patient was taken up for surgery with due consent. Specimen excised was a solitary 6 X 6 X 3.5 cm with gray-white firm homogenous firm lesion. Microscopy revealed sheets of histiocytes with ovoid, round nuclei and abundant cytoplasm, some of which contained lipid. Nodular collections of small lymphocytes and plasma cells were noted. Some of the histiocytes showed haemophagocytosis and emperipolesis. Immunohistochemistry showed cells positive S-100, CD 68 and negative for CD1a, SMA, CK. Desmin and a final diagnosis of extra nodal RosaiDorfman Disease was confirmed.
Discussion
Rosai-Dorman Disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, was originally described by Destombes in 1965 [1]. Subsequently, it was characterized as a distinct clinicopathologic disorder in 1969 by Rosai and Dorfman [2]. It is a benign self-limiting histiocytic proliferative disorder of unknown aetiology, which usually occurs in the first two decades of life. It is believed to be a reactive process and infection or an undefined immunological defect initiated by some other organism is believed to be responsible for the disease. RDD usually presents with painless massive cervical lymphadenopathy. Extra nodal involvement is common and is found mainly in the head and neck regions [3]. RDD can also present in any extra nodal site, with common sites including the skin and soft tissue, the Central Nervous System (CNS), and, less commonly, the gastrointestinal tract [3]. Cutaneous Rosai-Dorfman Disease (CRDD) is a rare proliferative disorder of histiocytes with unknown etiology, broadly different from systemic Rosai-Dorfman disease. The Cutaneous-only form of RDD (CRDD) is a clinically distinct entity from RDD and some researchers suggest that it may be a different clinicopathological entity from nodal RDD. In reported cases of CRDD, patients with CRDD are 45 years older compared with patients who have RDD. Women with CRDD appear to be more affected than men and most cases have been seen among Caucasian and Asian populations [4]. In CRDD, patients typically present with normal laboratory data and no adenopathy. Lesions in CRDD can vary, ranging from less than 1 cm to 30 cm or more at their greatest dimensions. Multiple lesions are generally present and are typically red-brown papules or nodules. The most common site of skin involvement is the torso followed by the head and neck region [5]. Most patients with CRDD follow a benign clinical course, with a frequent and spontaneous resolution of lesions. FNAC is a useful and reliable tool for the diagnosis of RDD, and as such, biopsy is avoidable [6]. Cytology usually reveals numerous large histiocytes with abundant pale cytoplasm and phagocytosed lymphocytes (emperipolesis). In emperipolesis, the lymphocytes are not attacked by enzymes and appear intact within the histiocytes (in contrast to phagocytosis). The phenomenon of emperipolesis is highly useful for the diagnosis of RDD using FNAC. Differential diagnoses, such as Langerhans Cell Histiocytosis (LCH), lymphoma and nonspecific sinus hyperplasia, lack lymphophagocytosis. In RDD, histiocytes are strongly positive for S-100 protein, negative for CD1a and variably positive for CD68. LCH is positive for both S-100 protein and CD1a. Our patient presented with a solitary painless swelling in the right para spinal area which was increasing in size and causing a lot of mental agony as she had been provisionally diagnosed as a neoplasm elsewhere and counseled for the same. Despite confirmation of diagnosis and reassurance about the non malignant nature of RDD, patient in order to allay her fear she wanted to get operated and reconfirm the diagnosis. RDD is self-limiting, 20% of cases show spontaneous regression without therapy [7]. Relapsing and remitting RDD without treatment has been reported in another 70% of patients in literature, thereby complicating the timing of when to use therapy. Patients requiring treatment, surgery is an appropriate option for disease that can be excised, including single nodal areas, primary CNS involvement, or localized primary RDD [7,8]. This was in our case. We have advised regular follow up for our patient. Radiotherapy, cryotherapy, topical chemotherapy as adjuvant therapy have been described in literature but with varying success [9,10].
Figure 1
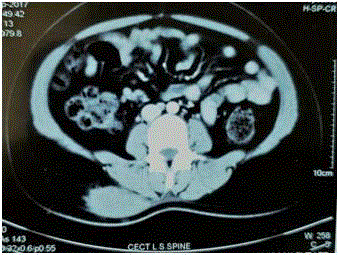
Figure 1
CECT LS SPINE Showing: 5.8 X 5.78 X 2.78 cm solitary
hyperdense and enhancing lesion in the subcutaneous plane in the right
posterior para spinal region without any extension or bony erosion or
calcification or necrosis.
Figure 2
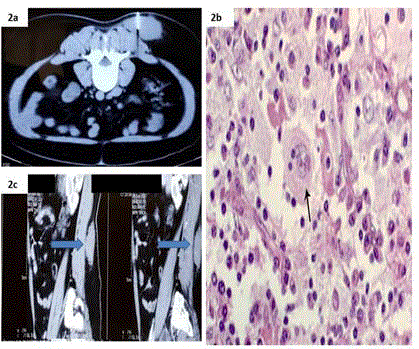
Figure 2
a: CECT LS spine showing FNAC needle in situ in the lesion.
b: Microscopy showed sheets of histiocytes with ovoid, round nuclei and
abundant cytoplasm, some of which contained lipid. Nodular collections of
small lymphocytes and plasma cells were noted. c: CECT LS SPINE showing
vertical extent of lesion.
Conclusion
Rosai-Dorfman disease is a histiocytic disorder with no malignant potential that classically presents with massive, painless cervical lymphadenopathy, fever, and an elevated erythrocyte sedimentation rate. Common extra nodal sites include the skin and the central nervous system. A high degree of clinical suspicion is needed to make the diagnosis because the differential diagnosis includes both malignancy and other histiocytic disorders. For patients who require therapy, surgical resection is the mainstay of treatment. Health care professionals are requested to explain and counsel patients for the same.
References
- Destombes P. [Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali. (4 cases)]. Bull Soc Pathol Exot Filiales. 1965;58(6):1169-75.
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol.1969;87(1):63-70.
- Sandoval-Sus JD, Sandoval-Leon AC, Chapman JR, Velazquez-Vega J, Borja MJ, Rosenberg S, et al. Rosai-Dorfman disease of the central nervous system: report of 6 cases and review of the literature. Medicine (Baltimore). 2014;93(3):165-75.
- Picarsic J, Jaffe R. Nosology and pathology of langerhans cell histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):799-823.
- Brenn T, Calonje E, Granter SR, Leonard N, Grayson W, Fletcher CD, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24(5):385-91.
- Das DK, Gulati A, Bhatt NC, Sethi GR. Sinus histiocytosis with massive lymphadenopathy (Roasi-Dorfman disease): report of two cases with fine needle aspiration cytology. Diagn Cytopathol. 2001;24(1):42-5.
- Lima FB, Barcelos PS, Constâncio AP, Nogueira CD, Melo-Filho AA. Rosai-Dorfman disease with spontaneous resolution: case report of a child. Rev Bras Hematol Hemoter. 2011;33(4):312-4.
- Forest F, N’Guyen AT, Fesselet J, Metellus P, Bouvier C, de Paula AM, et al. Meningeal Rosai-Dorfmandisease mimicking meningioma. Ann Hematol. 2014;93(6):937-40.
- Dalia S, Sagatys E, Sokol L, Kubal T. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21(4):322-7.
- Wang KH, Chen WY, Liu HN, Huang CC, Lee WR, Hu CH. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154(2):277-86.