Review Article
The Promising Role of Let-7 Microrna in Colorectal Cancer: Practical Points for Clinicians
Nikolaos Margetis1,2*, Athanassios Kotsinas2,3 and Theodoros Mariolis-Sapsakos4,5
1Athens Euroclinic, Department of Gastroenterology, University of Athens, Greece
2Molecular Carcinogenesis Group, Laboratory of Histology and Embryology, Medical School, University of Athens, Greece
3Assistant Professor, Laboratory of Histology and Embryology, Medical School, University of Athens, Greece
4Aghioi Anargyroi” Hospital, Athens, Greece
5Assistant Professor, Laboratory of Anatomy, School of Nursing, University of Athens, Greece
*Corresponding author: Nikolaos Margetis, Athens Euroclinic, Department of Gastroenterology, University of Athens, Greece
Published: 02 Jan, 2018
Cite this article as: Margetis N, Kotsinas A, Mariolis-
Sapsakos T. The Promising Role of
Let-7 Microrna in Colorectal Cancer:
Practical Points for Clinicians. Clin
Oncol. 2018; 3: 1390.
Abstract
Colorectal cancer, one of the most common cancers, displays disproportionally high mortality,
taking into consideration the enormous amount of data collected over the recent decades and the
broad use of preventive colonoscopy worldwide. MicroRNAs, small, non coding RNA molecules,
regulate many critical steps of the entire stages of colorectal tumorigenesis process and shed light to
the in depth comprehension of the complex genetic environment that governs the process. Let-7 is
the largest microRNA family studied and consists of ten mature members, which exhibit redundancy
in colorectum. Its main role is to promote differentiation and depress stemness, both in normal and
neoplastic colon. It represses or abrogates translation of other genes, by complementary binding to
their mRNAs. It is mostly considered a tumor-suppressor, as it targets mainly oncogenes; among
them K-ras is the dominant. Let-7 establishes feedback loops with the majority of its targets. Its
expression levels increase over fetus development, are higher on the top vs the bottom of the colonic
crypt and are downregulated, as normal colorectal epithelium progress to neoplasia. Although let-
7’s tumor-suppressive effect is dependent on the primitive or advanced stage of colorectal neoplasia,
colorectal cancer tissues’ let-7 levels are proportionate to cancer stage. High let-7 levels may prove a
favorable prognostic and predictive biomarker. In case scientists overcome multiple limitations, the
administration of mature let-7 inside colorectal cancer tissues (directly, delivering synthetic let-7
or indirectly, using a viral vector), may improve future management of CRC, since underexpressed
let-7 levels favor every single stage of colonic oncogenic transformation.
Keywords: Let-7; K-ras mutation; LCS6; Biomarker; Colorectal cancer
Abbreviations
APC: Adenomatous Polyposis Coli protein
CDKs: Cyclin-dependent kinases
CRC: Colorectal Cancer
DDR: DNA damage response
DSBs: DNA double-stand breaks
EMT: Epithelial-Mesenchymal Transition
GSK-3b: glycogen synthase kinase 3b
FFPE: formalin-fixed, paraffin-embedded
HMGA2: High Motility Group A2
IGF2BP1: Igf2 mRNA binding protein 1
Let-7: lethal-7 microRNA
LCSs: let-7 complementary sites
MAPK: the pathway of MAP kinases
miRNA: MicroRNA
NFκΒ: Nuclear factor of κ light polypeptide gene enhancer in
B-cells
PDH-K: Pyruvate dehydrogenase kinase
PDK1: Phosho-inositide-dependent kinase 1
PI3K: Phosphatidylinositole 3-kinase
RAS-GDP: the inactive form of oncogene RAS
RAS-GTP: the active form of oncogene RAS
RISC: RNA-induced silencing complex
SNP: Single Nucleotide Polymorphism
TGF-β: transforming growth factor-β
TGF-β Rec: cell membrane receptor of TGF-β
UTR: UnTranslated Region, Wnt: Wnt signaling pathway
Introduction
Colorectal cancer is the third leading cause of cancer-related deaths on the USA [1]. Colorectal carcinogenesis is governed by the interaction between the inherited genome, the somatic genetic alterations and environmental factors [2]. MicroRNAs consist of 18- 25 nucleotides and are crucial epigenetic regulators of the entirety of steps of colorectal carcinogenesis process [3-6]. They don’t translate into proteins; instead, they downregulate the translation process of their mRNA-target [7]. Let-7 family, the best miRNA family studied [8], consists of ten mature members [9]. Let-7 forms RISC [7] and guides it to the 3΄ UTR of its mRNA-target: partial complementarity represses mRNA translation, whereas high complementarity degrades mRNA [10].
The Increasing Number of mRNA Targets of Let-7
Mature let-7 is broadly viewed a tumor-suppressor gene,
since the majority of its targets are strong oncoproteins [11-13]. It
downregulates a plethora of mRNAs, mainly participating in the
two major initiating oncogenic pathways in CRC (MAPK and Wnt).
Furthermore, it targets other oncogenes, as c-myc [8,13], HMGA2
[14] and LIN28 A/B [8-13]). Lin28, a RNA-binding protein, promotes
stemness [12,15] and inhibits differentiation [16,17]. When Lin28
is downregulated, Wnt signaling is hypoactivated and β-catenin
levels are lowered [8-13,15-17]. β-catenin links Wnt and MAPK
pathways, both directly, achieving mutual enhancement with RASGTP
state and indirectly, promoting GSK-3b [18]. Therefore, let-7
downregulates MAPK pathway in a dual manner, by destabilizing
RAS into RAS-GDP state and by lowering levels of β-catenin
cytoplasmic pool (Figure 1).
The dominant target of let-7 is K-ras, one of the earliest mutations
in colorectal carcinogenesis process [18]. Let-7 represses K-ras
protooncogene [19-30] and K-ras oncogene [23-30] through ten
different LCSs, located in the 3΄ UTR of K-ras mRNA [31]. Let-7
silences K-ras mRNA only in case LC6 is intact. LCS1 and, mostly
LCS6, are well-studied in CRC. A SNP variant in LCS6, a germ-line
functional variation of T to G substitution, namely G-allele [20-31],
is found in 6% of the population worldwide [25] and diminishes the
binding affinity of let-7 with K-ras [30].
In wild-type K-ras CRCs, G-allele carriers (TG or GG) have
lower levels of let-7. Therefore, G-allele reinforces the tumorigenic
effect of low let-7 levels and is currently viewed as an inheritable
tumor-promoting alteration [20]. A clonal selection inside the
growing neoplasm favoring more aggressive clones, which bear
both the hostle germline variant and the oncogene-promoting low
let-7 levels is presumed [23,25], probably accomplished through the
use-it-or-lose-it mechanism [20]. The ultimate result is the gradual
increase of the G-allele throughout the successive stages of colorectal
carcinogenesis [25]. Despite the striking differences among research
works [21-30,32], the collective data imply that the interplay among
three independent factors (1 K-ras status 2 let-7’s levels and 3 LCS6’s
genotype) determines the course and prognosis of adenomatous
colorectal neoplasias.
The arsenal of let-7 targets is completed by a plethora of
oncogenes. Let-7 downregulates PI3K/AKT pathway, targeting
protein AKT, mTOR and PDK1[8]. It prevents dissemination of CRC
cells by inhibiting IGF2BP1 [16], IFG1R and PDH-K [8]. It depresses
the progress of cell cycle, as it targets cyclins (A,D1,D2,D3) and CDKs
(2,4,6,25A,34A) [8,13,33]. It inhibits the antiapoptotic protein BCLXL,
enhancing apoptosis [8]. Last, it represses IL-6 and STAT-3,
which are indispensable for the transition of inflammation to CRC
[8,34].
Nevertheless, let-7 is capable to exhibit oncogenic properties
as well. High levels of let-7 limit apoptosis, by inhibiting the death
receptor Fas [35] and promote proliferation, by downregulating
the antiproliferative TGF-β [10]. Let-7 suppresses innate immune
reactions against CRC by inhibition of Toll-like receptor 4 [36], by
inhibition of NFκΒ pathway [8] and by targeting mTOR RNA [37].
Last, let-7 may repress the translation of TP53 gene [38].
The interplay between let-7 and its effectors is complex; nearly
all let-7 targets behave as its reciprocal regulators, negatively in their
majority. Ras negatively regulates let-7 by activation of NFκΒ [34]
and by upregulating LIN28 via MAPK activated c-myc expression
[8]. NFκΒ fosters let-7a expression by inducing its promoter [39].
Let-7 establishes a negative feedback loop with LIN28A/B [8,13,17],
with HMGA2 [8,14] and with c-myc [8,35,40,41]. Wnt pathway
activation increases β-catenin levels, which hyperactivates LIN28
[17]; both repress let-7. p53 protein suppresses let-7: in early stages
of tumorigenesis wtp53 inhibits let-7 (directly, by binding to its
promoter [42,43]) and indirectly, by inhibiting Fas [43]), whereas
in late stages of CRC, mutated p53 suppresses let-7 by inhibiting its
maturation process [44]). Last, but not least, let-7 targets itself; it is
positively autoregulated, (mature let-7 enhances its own biosynthesis
[45]) and is negatively autoregulated (it drives its own degradation
in case it does not fulfill its pursuit, through the use-it-or-lose-it
mechanism) [20] (Figure 1).
The Evolution of Let-7’s Colorectal Levels from Early Fetal Life to Late Carcinogenesis
Mature let-7 is undetectable in early fetal life, whereas let-7’s
expression increases during late embryogenesis [11]. In embryonic
and in adult life, let-7’s major role is to promote differentiation: it
is undetectable in embryonic [12] and in normal colon stem cells
[11,13,17], whereas higher levels are maintained in embryonic and
adult differentiated cells [11,12] and tissues [13].
Let-7 levels display striking inhomogeneity both in normal and
neoplastic colon; its global mission is to establish obstacle against
stemness. Being upregulated in stem cells, it forces and governs their
differentiation [11], it controls the timing of differentiation and is
upregulated upon differentiation [46]. Acting upon differentiated
cells, it exerts a dual effect: first, it prevents their dedifferentiation
to stem cells [46] and, second, it represses cell cycle progression and
therefore halts their proliferation as they migrate towards the top of
the crypt, thus preventing their neoplastic transformation [47]. Let-
7 keeps differentiated cells differentiated; when this function fails,
neoplastic conformation initiates [10]. As CRC is a disease of stem
cells [48,49], the tumor-suppressive role of let-7 is explained by its
action upon cells of the top and of the bottom of the crypt. A gradual
increase of let-7 levels is thereby evident across normal crypt axis [48]:
normal colorectal stem cells in the basis of the crypt express low or
undetectable let-7 levels [11,13,17,47], in contrast to increasingly high
let-7 content in the differentiated cells in the transition and the villous
domain of the crypt [11]. Let-7 is the most representative marker of
epithelial differentiation in colon [47,50].
Colonic neoplastic transformation, a process similar to reversed
embryogenesis [11], presupposes loss of let-7 even from its very
early stages [50]. Indeed, mature let-7 levels are underexpressed in
colorectal neoplastic tissues compared to normal adjacent tissues
both in premature stage (benign adenoma) and late stage (carcinoma)
of the process (Figure 2) [44].
Similarly, upregulation of let-7 guides colorectal cancer stem cell
transition to differentiated cancer cells [11]. Stage III/IV CRC tissues
bear higher levels of let-7-a/let-7-b compared to their corresponding
stage I/II [36]. Mature let-7-a levels parallels the progression of
colorectal cancer [51] and let-7 exhibits increased levels in advanced
colorectal cancer tissues [52-54]. In case metastases occur, let-7-a
continues to increase its expression [51]. The gradual increase of the
tumor-suppressor let-7 through the successive stages of CRC may
reflect either the pressure of natural selection, or the under-defined
role of tumor-promoting properties of let-7. Last, inflammatory
stroma surrounding cancerous cells harbors up to 4 times higher let-
7 levels compared to their paired cancer cells [36], implying that let-7
may regulate the stroma/cancer cells interaction inside the growing
colorectal neoplasia (Figure 2).
Figure 1
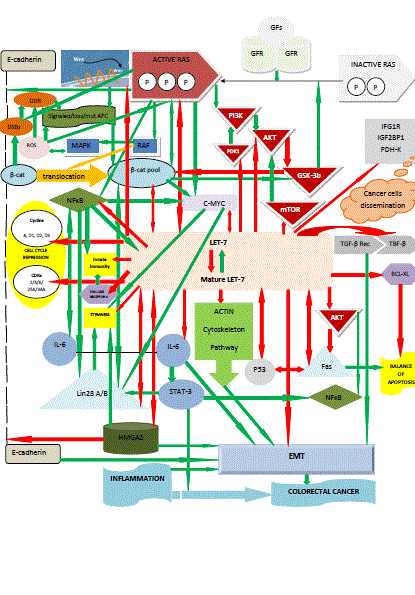
Figure 1
The tumor-suppressive role of let-7 in colorectal cancer: let-7 holds a central regulatory position in the complicated cross-talk between the major pathways
of colorectal tumorigenesis process; it is interdependent on multiple effectors and inhibits cell cycle, innate immunity against cancer cells, epithelial-mesenchymal
transition, dissemination of transformed cells and stemness. The arrows filled with red colour indicate inhibition of the target they direct; the arrows filled with green
colour indicate promotion of the target they direct (based on references 8, 10, 13, 14, 16-30, 32, 34-40, 42-45, 58-60, 65 and 87-94).
The Decalogue of let-7’s Action in Colorectum
1. Mature let-7 members, though don’t harbor absolutely the
same sequence, they greatly resemble one another [45]. They share
identical seed sequence, crucial for target recognition [55], which
ensures that their actions overlap, both in normal and neoplastic
colon [56].
2. Let-7 prevents all the hallmarks of cancer [57]. Low let-7 levels
force growing cells to sustain proliferation signaling and to evade
tumor suppressors [8,10], enhance EMT [8], provoke the transition
of inflammation to cancer [31,34] and promote genomic instability,
mainly by K-ras hyperactivation, which causes DSBs and DDR [58-
60], (Figure 1).
3. Despite the relative abundance of let-7 in colon [5], no
absolutely determined normal levels of let-7 expression in colorectum
have been evaluated so far. Let-7 levels are quantified comparatively,
in relation to its own expression in different situations, spatially [61]
or temporally [11].
4. Let-7 isoforms aren’t equally expressed both in normal and in
the neoplastic colon. First, let-7-a, let-7-b [56], let-7-c [47], let-7-g
and let-7-f [56] predominate in normal colonic epithelia. Second,
let-7a and let-7-b are the prevalent isoforms found inside colorectal
cancer cell lines [62]. Third, let-7-a [63,64], let-7-b [56,65], let-7-c/
let-7-f [65] levels are preferentially repressed in colorectal neoplastic
tissues compared to their paired normal ones.
5. Let-7’s action is tissue-specific, disease-specific and cellspecific
[46]. Acting upon stem cells, it forces them to differentiate
[46], whereas it blocks the proliferation [47] and prevents the
dedifferentiation of differentiated cells [46].
6. Let-7’s action in a given cell of the colonic epithelium in a
given time depends on the relative intracellular concentrations of its
positive and negative regulators [8,13-17,34,39-44].
7. Let-7’s expression in a given cell of the colonic epithelium
in a given time depends on the relative luminal concentrations of
environmental factors, as several dietary components upregulate (e.g.
spinach [66]) or downregulate (e.g. polyamines [67]) mature let-7.
8. The antitumorigenic or oncogenic effect of let-7 depends on the
stage of the tumorigenesis process. In the stage of colorectal adenoma
and early carcinoma, low let-7 levels are beneficial, i.e. they halt the
process: low let-7 levels induce apoptosis [35,37,43], induce oncogeneinduced
senescence [8,22], mainly via K-ras hyperactivation [18,68]
and enhance innate immunity against cancer progression [8,36].
In late carcinoma stages, in the context of mutated p53, low let-7
inhibits apoptosis [8,35,37] and increase stemness [46]. Thereby, in
the advanced CRC stages, low let-7 levels are deleterious.
9. The antitumorigenic or tumorigenic effect of let-7 depends
on the cell inside the growing colorectal tumor where it exerts its
action. Increased aggressiveness of CRC is induced by low let-7 levels
inside colorectal cancer cells (they drive stemness, EMT, invasion
and metastasis [8,45,50,69]) and by high let-7 levels inside stroma
cells (they lead to diminished lymphocytic immunity against cancer
cells [36]). The relative content of different cell types inside the tumor
creates the dominant cellular environment and predetermines the
overall contribution of let-7 in tumor’s behavior.
10. Underexpressed let-7 accelerates every single stage of
colorectal tumorigenesis [70]. In the incipient steps, low let-7 levels
activate Wnt pathway, via the upregulation of β-catenin and c-myc
[8,14,17,18,35,40]. Low let-7 levels activate K-ras and AKT in the
stage of EGFR signaling [8, 17, 21-30] and TGF-β response in the next
stage [8,10]. Following this, low let-7 levels permit p53 protein to over
express [38]. Finally, low let-7 levels enhance the cancer-promoting
EMT, as let-7 downregulates independently four EMT-favoring
factors, i.e. TGF-β [8,10], HMGA2 [74], IL-6 [8,34] and the acting
cytoskeleton pathway [8] (Figure 1).
Figure 2
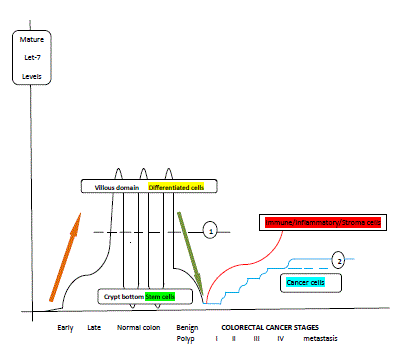
Figure 2
The suggested relative concentrations of mature let-7 in colorectal
cells over the phases of colon evolution: fetal colon, normal adult colon,
neoplastic adult colon. Let-7 levels in colon mucosa increase gradually
during development of fetus and reach approximately the average levels of
the adult colon mucosa (line 1) after birth. A gradient is established from the
bottom of the crypt (low or undetectable let-7 levels) to the villous domain
on the top of the crypt (high let-7 levels) across the adult colon crypt axis.
Colorectal adenoma cells bear intermediate let-7 levels i.e. levels between
these of normal colorectal mucosa (level 1) and those found on the first
stage of colorectal cancer. Colorectal cancer stages display incremental
levels of let-7 from stage I to stage IV, and probably from stage IV to
metastatic disease (blue curved line). Nevertheless, the maximum levels
of let-7 in colorectal cancer cells (line 2) are maintained below the average
normal colorectal mucosal levels (line 1). Stroma cells surrounding cancer
contain increased let-7 levels (red curved line), compared to their paired
cancerous colorectal cancer cells. According to the progress of let-7 levels,
colorectal carcinogenesis process (green arrow) is indeed the inversion of
the embryogenesis process (brown arrow) (based on references 12, 13, 17,
36, 43, 45-55, 61, 69, 71 and 76).
The Clinical Role of Let-7 as A Biomarker in Colorectal Cancer
Let-7 is very stable molecule in extreme pH values and in boiling,
it is easy to be extracted from fresh or archival FFPE tissues, from
stool or from blood/serum and can resist degradation over time. It
is therefore candidate for being used as a biomarker [71,72]. Clinical
research has given conflicting effects regarding the clinical value
of let-7 in CRC. Concerning its diagnostic role, research works
have shown either upregulated [73,74] or downregulated [75] let-
7 isoforms levels in blood circulation of CRC patients compared to
control group. Regarding prognosis, poor prognosis was correlated
either to low let-7 levels [32,56] or to increased let-7 [37,76,77] in
CRC tissues. Similarly, two investigators studying the influence of
let-7 on survival of CRC patients, generated conclusions in opposite
directions [78,79]. Last, regarding its predictive role to non-surgical
therapy of CRC, most (but not all) studies have shown that high let-
7 content in CRC tissues are correlated to radiosensitivity [8,55,80],
to sensitivity to chemotherapeutics [5,54,55,78,79] and to improved
prognosis after anti-EGFR antibodies (cetuximab) administration
[22,32]. To make things complicated, mounting evidence suggests that
increased neoplastic let-7 levels, a presumably favorable predictive
factor, which might drive doctors to recruit patients for advanced
therapy, are downregulated during the corresponding therapeutic
modality, i.e. CRC is capable to resist to radiotherapy [42,55,80] and
to chemotherapy [81].
Finally, studies have not associated G-allele to CRC development
[28]. Surprisingly, some works showed that it improves prognosis
[25], whereas others demonstrated neutral effect [26]. Regarding its
predictive role, a few works demonstrated that, despite its tumorigenic
properties, G-allele was associated with improved prognosis both
in naïve patients and in patients after chemotherapy or anti-EGFR
antibodies therapy. Nevertheless, G-allele was not proved to be an
independent predicting factor for CRC patients receiving anti-EGFR
antibodies or other advanced therapy [21,22,25,27].
The Involvement of Let-7 in Colorectal Cancer Therapy
Let-7 holds dual properties; hence let-7-based therapy may be
directed analogously. In tissues where let-7’s tumor-promoting
capabilities dominate, our therapeutic target is its inhibition. This
is achieved by anti-sense oligonucleotides (ASOS), anti-miRNA
oligonucleotides (AMOS) or microRNA sponges [10,82,83].
Nevertheless, let-7 is largely a tumor-suppressor in CRC; therefore
our main pursue is to rocket up its expression or to restore its levels
(gene therapy). To deliver let-7, adenoviral [10] or retroviral based
[46] vectors have been proposed. Moreover, synthetic let-7 members,
chemically modified RNA molecules that mimic mature isoforms’
action have been invented and are delivered directly inside colorectal
cancer tissue [46]. Indeed, delivering let-7 in CRC animal models has
given encouraging results [46].
Unfortunately, a plethora of limitations and obstacles renders the
manipulation of let-7 in CRC problematic. First, it is not feasible to
decide whether to increase or lower let-7 levels in a given CRC, since
low let-7 levels are not the characteristic of every single colorectal
cancer tissue [46]. In spite the fact that the majority of studies have
demonstrated reduction of CRC growth and invasion after restoration
of let-7 levels, experimental data have given contradictory results
[17,56,58]. As in vivo genetic studies of let-7 function are lacking [84],
we are unaware about the isofrom(s) of let-7 that need restoration
in a given colorectal neoplasm. The effects of exogenous let-7 on
regulatory mechanisms of endogenous let-7 maturation and the
ultimate effect of their interaction in the carcinogenesis process are
largely obscure. Besides this, many critical genetic driver mutations
are necessary for CRC development [85,86] and the isolated effect of a
manipulator with central role, as let-7, upon them is largely unknown.
Additionally, the possibility of immunization against exogenous let-7
has not been elucidated and the optimal delivery system is a matter
of intense research [46]. The inhomogeneity of let-7 inside colorectal
tissue renders almost impossible to achieve cellular target specificity,
i.e. to restore let-7 levels exclusively in cancerous cells and not in
stroma cells [10,12,83]. Moreover, since low let-7 levels are beneficial
in early CRC stages, it is unknown whether the anti-tumor effect of
increased let-7 levels is limited to advanced stages of tumorigenesis.
Furthermore, synthesis and purification of therapeutic let-7 is quite
difficult and let-7 restoration methods are not yet satisfactory [10].
The last hurdle to overcome is skin toxicity, attributed to virus-based
let-7 delivery [32].
Let-7 and the Forthcoming Therapy of CRC: Prospects and Proposals
To put in a nutshell, let-7 is a strong manipulator of every single intermediate stage of colorectal tumorigenesis, inhibiting multiple crucial effectors. Apart from clarifying its diagnostic, prognostic and predictive role in CRC, future efforts should concentrate on let-7’s main properties, i.e. the promotion of differentiation and the abolishment of stemness. Gene therapy, restoring let-7 levels or forcing its expression, may become a real fact, if we fully understand let-7’s biology, regulation and interdependencies in normal and neoplastic colon before and after exogenous let-7 delivery, if we determine which genes are predominantly influenced when let-7 is delivered inside CRC tissue and if we shed light to the regulation and function of normal and CRC stem cells. Important issues should be resolved: ineffective delivery, difficulty in transducing large volume of cells in the tumors [46], inability to determine the “let-7 identity” of a given tumor and incapability to target let-7 delivery in a specific cell group inside the tumor. Furthermore, in order to use let-7 as an in vivo manipulator, the therapeutic indications (which CRC patients, which stage), the putative combination with other advanced therapies, the ultimate goals (remission of preexisting tumors vs prevention of tumor initiation vs halting neoplasia progression), specific details (which let- 7 isoforms are necessary to deliver) and the contraindications of let-7 delivering strategy should become definite. Moreover, the undesired effects of let-7 administration (skin toxicity, induction of deleterious immune activation) must be determined and overcome. Finally, in order to be realistic, novel, let-7-based, therapeutic patents, like those recently invented in China and Australia [46] should focus on the field of CRC therapeutics.
References
- Siegel R, Desantis C, Jemal A. Colorectal cancer statistics. CA Cancer J Clin. 2014;64(2):104-17.
- Worthley DL, Whitehall VL, Spring KJ, Leggett BA. Colorectal carcinogenesis: road maps to cancer. World J Gastroenterol. 2007;13(28):3784-91.
- Lizarbe MA, Calle-Espinosa J, Fernández-Lizarbe E, Fernández-Lizarbe S, Robles MÁ, Olmo N, et al. Colorectal Cancer: From the Genetic Model to Posttranscriptional Regulation by Noncoding RNAs. Biomed Res Int. 2017;7354260.
- Rahmani F, Avan A, Hashemy SI, Hassanian SM. Role of Wnt/β-catenin signaling regulatory microRNAs in the pathogenesis of colorectal cancer. J Cell Physiol. 2018;233(2)811-7.
- Strubberg AM, Madison BB. MicroRNAs in the etiology of colorectal cancer: pathways and clinical implications. Dis Model Mech. 2017;10:197-214.
- Guo Y, Bao Y, Yang W. Regulatory miRNAs in Colorectal Carcinogenesis and Metastasis. Int J Mol Sci. 2017;18(4):E890.
- Macfarlane LA, Murphy PR. MicroRNA: Biogenesis, Function and Role in Cancer. Curr Genomics. 2010;11(7):537-61.
- Wang T, Wang G, Hao D, Liu X, Wang D, Ning N, et al. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol Cancer. 2015;14:125.
- Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008; 18 (10):505-16.
- Barh D, Malhotra R, Ravi B, Sindhurani P. MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010;17(1):70-80.
- Büssing I, Slack FJ, Grosshans H. let-7 microRNAs in development, stem cells and cancer. Trends Mol Med. 2008;14(9):400-9.
- Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17(1):F19-36.
- Su JL, Chen PS, Johansson G, Kuo ML. Function and regulation of let-7 family microRNAs. Microrna. 2012;1(1):34-9.
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 21. 2007;21(9):1025-30.
- Pang M, Wu G, Hou X, Hou N, Liang L, Jia G, et al. LIN28B promotes colon cancer migration and recurrence. PLoS One. 2014;9(10):e109169.
- Hamilton KE, Noubissi FK, Katti PS, Hahn CM, Davey SR, Lundsmith ET, et al. IMP1 promotes tumor growth, dissemination and a tumor-initiating cell phenotype in colorectal cancer cell xenografts. Carcinogenesis. 2013;34(11):2647-54.
- Tu HC, Schwitalla S, Qian Z, LaPier GS, Yermalovich A, Ku YC, et al. LIN28 cooperates with WNT signaling to drive invasive intestinal and colorectal adenocarcinoma in mice and humans. Genes Dev 29. 2015;29(10):1074-86.
- Margetis N, Kouloukoussa M, Pavlou K, Vrakas S, Mariolis-Sapsakos T. K-ras Mutations as the Earliest Driving Force in a Subset of Colorectal Carcinomas. In Vivo. 2017;31(4):527-42.
- Bueno MJ, Pérez de Castro I, Malumbres M. Control of cell proliferation pathways by microRNAs. Cell Cycle. 2008;7(20):3143-8.
- Crowley EH, Arena S, Lamba S, Di Nicolantonio F, Bardelli A. Targeted knock-in of the polymorphism rs61764370 does not affect KRAS expression but reduces let-7 levels. Hum Mutat. 2013;35(2):208-14.
- Saridaki Z, Weidhaas JB, Lenz HJ, Laurent-Puig P, Jacobs B, De Schutter J, et al. A let-7 microRNA-binding site polymorphism in KRAS predicts improved outcome in patients with metastatic colorectal cancer treated with salvage cetuximab/panitumumab monotherapy. Clin Cancer Res. 2014;20(17):4499-510.
- Zhang W, Winder T, Ning Y, Pohl A, Yang D, Kahn M, et al. A let-7 microRNA-binding site polymorphism in 3'-untranslated region of KRAS gene predicts response in wild-type KRAS patients with metastatic colorectal cancer treated with cetuximab monotherapy. Ann Oncol. 2011;22(1):104-9.
- Graziano F, Canestrari E, Loupakis F, Ruzzo A, Galluccio N, Santini D, et al. Genetic modulation of the Let-7 microRNA binding to KRAS 3'-untranslated region and survival of metastatic colorectal cancer patients treated with salvage cetuximab-irinotecan. Pharmacogenomics J. 2010;10(5):458-64.
- Sebio A, Paré L, Páez D, Salazar J, González A, Sala N, et al. The LCS6 polymorphism in the binding site of let-7 microRNA to the KRAS 3'-untranslated region: its role in the efficacy of anti-EGFR-based therapy in metastatic colorectal cancer patients. Pharmacogenet Genomics. 2013;23(3):142-7.
- Smits KM, Paranjape T, Nallur S, Wouters KA, Weijenberg MP, Schouten LJ, et al. A let-7 microRNA SNP in the KRAS 3'UTR is prognostic in early-stage colorectal cancer. Clin Cancer Res. 2011;17 (24):7723-31.
- Ryan BM, Robles AI, Harris CC. KRAS-LCS6 genotype as a prognostic marker in early-stage CRC-letter. Clin Cancer Res. 2012;18:3487-8.
- Langevin SM, Christensen BC. Let-7 microRNA-binding-site polymorphism in the 3'UTR of KRAS and colorectal cancer outcome: a systematic review and meta-analysis. Cancer Med. 2014;3(5):1385-95.
- Kjersem JB, Ikdahl T, Guren T, Skovlund E, Sorbye H, Hamfjord J, et al. Let-7 miRNA-binding site polymorphism in the KRAS 3'UTR; colorectal cancer screening population prevalence and influence on clinical outcome in patients with metastatic colorectal cancer treated with 5-fluorouracil and oxaliplatin +/- cetuximab. BMC Cancer. 2012;12:534.
- Sha D, Lee AM, Shi Q, Alberts SR, Sargent DJ, Sinicrope FA, et al. Association study of the let-7 miRNA-complementary site variant in the 3' untranslated region of the KRAS gene in stage III colon cancer (NCCTG N0147 Clinical Trial). Clin Cancer Res. 2014;20(12):3319-27.
- Sclafani F, Chau I, Cunningham D, Peckitt C, Lampis A, Hahne JC. Prognostic role of the LCS6 KRAS variant in locally advanced rectal cancer: results of the EXPERT-C trial. Ann Oncol. 2015;26(9):1936-41.
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635-47.
- Ruzzo A, Graziano F, Vincenzi B, Canestrari E, Perrone G, Galluccio N, et al. High let-7a microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapy-refractory metastatic disease. Oncologist. 2012;17(6):823-9.
- Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim Biophys Acta. 2011;1812(5):592-601.
- Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139(4):693-706.
- Geng L, Zhu B, Dai BH, Sui CJ, Xu F, Kan T, et al. A let-7/Fas double-negative feedback loop regulates human colon carcinoma cells sensitivity to Fas-related apoptosis. Biochem Biophys Res Commun. 2011;408(3):494-9.
- Dou R, Nishihara R, Cao Y, Hamada T, Mima K, Masuda A, et al. MicroRNA let-7, T Cells, and Patient Survival in Colorectal Cancer. Cancer Immunol Res. 2016;4(11):927-35.
- Liu M, Chen H. The role of microRNAs in colorectal cancer. J Genet Genomics. 2010;37(6):347-58.
- Boominathan L. The tumor suppressors p53, p63, and p73 are regulators of microRNA processing complex. PLoS One. 2010;5(5):e10615.
- Wang DJ, Legesse-Miller A, Johnson EL, Coller HA. Regulation of the let-7a-3 promoter by NF-κB. PLoS One. 2012;7(2):e31240.
- Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci USA. 2009;106(9):3384-9.
- Wang Z, Lin S, Li JJ, Xu Z, Yao H, Zhu X, et al. MYC protein inhibits transcription of the microRNA cluster MC-let-7a-1~let-7d via non canonical E-box. J Biol Chem. 2011;286(46):39703-14.
- Saleh AD, Savage JE, Cao L, Soule BP, Ly D, DeGraff W, et al. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on p53. PLoS One. 2011;6(10):e24429.
- Hau A, Ceppi P, Peter ME. CD95 is part of a let-7/p53/miR-34 regulatory network. PLoS One. 2012;7(11):e49636.
- Lee JY, Kim HJ, Yoon NA, Lee WH, Min YJ, Ko BK, et al. Tumor suppressor p53 plays a key role in induction of both tristetraprolin and let-7 in human cancer cells. Nucleic Acids Res. 2013;41(11):5614-25.
- Pasquinelli AE. The primary target of let-7 microRNA. Biochem Soc Trans. 2013;41(4):821-4.
- Chiu SC, Chung HY, Cho DY, Chan TM, Liu MC, Huang HM et al. Therapeutic potential of microRNA let-7: tumor suppression or impeding normal stemness. Cell Transplant. 2014;23(4-5):459-69.
- Madison BB, Liu Q, Zhong X, Hahn CM, Lin N, Emmett MJ, et al. LIN28B promotes growth and tumorigenesis of the intestinal epithelium via Let-7. Genes Dev. 2013;27(20):2233-45.
- Cui S, Chang PY. Current understanding concerning intestinal stem cells. World J Gastroenterol. 2016;22(31):7099-110.
- Zeki SS, Graham TA, Wright NA. Stem cells and their implications for colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8(2):90-100.
- Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle. 2009;8(6):843-52.
- Vickers MM, Bar J, Gorn-Hondermann I, Yarom N, Daneshmand M, Hanson JE, et al. Stage-dependent differential expression of microRNAs in colorectal cancer: potential role as markers of metastatic disease. Clin Exp Metastasis. 2012;29(2):123-32.
- Nam S, Kim B, Shin S, Lee S. MiRGator: an integrated system for functional annotation of microRNAs. Nucleic Acids Res 36 (Database issue). 2008;D159-64.
- Xi Y, Shalgi R, Fodstad O, Pilpel Y, Ju J. Differentially regulated micro-RNAs and actively translated messenger RNA transcripts by tumor suppressor p53 in colon cancer. Clin Cancer Res. 2006;12(7 Pt 1):2014-24.
- Nakajima G, Hayashi K, Xi Y, Kudo K, Uchida K, Takasaki K, et al. Non-coding MicroRNAs hsa-let-7g and hsa-miR-181b are Associated with Chemoresponse to S-1 in Colon Cancer. Cancer Genomics Proteomics. 2006;3(5):317-24.
- Kolenda T, Przybyła W, Teresiak A, Mackiewicz A, Lamperska KM. The mystery of let-7d - a small RNA with great power. Contemp Oncol (Pozn). 2014;18(5):293-301.
- King CE, Wang L, Winograd R, Madison BB, Mongroo PS, Johnstone CN, et al. LIN28B fosters colon cancer migration, invasion and transformation through let-7-dependent and independent mechanisms. Oncogene. 2011;30(40):4185-93.
- Hanahan, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74.
- Abulaiti A, Fikaris AJ, Tsygankova OM and Meinkoth JL. Ras induces chromosome instability and abrogation of the DNA damage response. Cancer Res 66. 2006;66(21):10505-12.
- Park MT, Kim MJ, Suh Y, Kim RK, Kim H, Lim EJ, et al. Novel signaling axis for ROS generation during K-Ras-induced cellular transformation. Cell Death Differ. 2014;21(8):1185-97.
- Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L, et al. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25(2):243-56.
- Akao Y, Nakagawa Y, Naoe T. Let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29(5):903-6.
- Liu H, D'Andrade P, Fulmer-Smentek S, Lorenzi P, Kohn KW, Weinstein JN, et al. mRNA and microRNA expression profiles of the NCI-60 integrated with drug activities. Mol Cancer Ther. 2010;9(5):1080-91.
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103(7):2257-61.
- Fang WJ, Lin CZ, Zhang HH, Qian J, Zhong L, Xu N. Detection of let-7a microRNA by real-time PCR in colorectal cancer: a single-centre experience from China. J Int Med Res. 2007;35(5):716-23.
- Michael MZ, O' Connor SM, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1(12):882-91.
- Parasramka MA, Dashwood WM, Wang R, Abdelli A, Bailey GS, Williams DE, et al. MicroRNA profiling of carcinogen-induced rat colon tumors and the influence of dietary spinach. Mol Nutr Food Res. 2012;56(8):1259-69.
- Paz EA, LaFleur B, Gerner EW. Polyamines are oncometabolites that regulate the LIN28/let-7 pathway in colorectal cancer cells. Mol Carcinog. 2014;53(1):E96-106.
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352-5.
- Ji J, Wang XW. A Yin-Yang balancing act of the lin28/let-7 link in tumorigenesis. J Hepatology. 2010;53(5):974-5.
- Slaby O, Svoboda M, Michalek J, Vyzula R. MicroRNAs in colorectal cancer. Translation of molecular biology into clinical application. Mol Cancer. 2009;8:102.
- Boisen MK, Dehlendorff C, Linnemann D, Schultz NA, Jensen BV, Høgdall EV et al. MicroRNA Expression in Formalin-fixed Paraffin-embedded Cancer Tissue: Identifying Reference MicroRNAs and Variability. BMC Cancer. 2015;15:1024.
- Xuan Y, Yang H, Zhao L, Lau WB, Lau B, Ren N et al. MicroRNAs in colorectal cancer: small molecules with big functions. Cancer Lett. 2015;360(2):89-105.
- Wang J, Huang SK, Zhao M, Yang M, Zhong JL, Gu YY, et al. Identification of a circulating microRNA signature for colorectal cancer detection. PLoS One. 2014;9(4):e87451.
- Ogata-Kawata H, Izumiya M, Kurioka D, Honma Y, Yamada Y, Furuta K, et al. Circulating exosomal microRNAs as biomarkers of colon cancer. PLoS One. 2014;9(4):e92921.
- Ghanbari R, Mosakhani N, Sarhadi VK, Armengol G, Nouraee N, Mohammadkhani A, et al. Simultaneous Underexpression of let-7a-5p and let-7f-5p microRNAs in Plasma and Stool Samples from Early Stage Colorectal Carcinoma. Biomark Cancer 7. 2016;7(1):39-48.
- Schepeler T, Reinert JT, Ostenfeld MS, Christensen LL, Silahtaroglu AN, et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008;68(15):6416-24.
- Mosakhani N, Lahti L, Borze I, Karjalainen-Lindsberg ML, Sundström J, Ristamäki R, et al. MicroRNA profiling predicts survival in anti-EGFR treated chemorefractory metastatic colorectal cancer patients with wild-type KRAS and BRAF. Cancer Genet. 2012;205(11):545-51.
- Dong Y, Wu WK, Wu CW, Sung JJ, Yu J, Ng SS. MicroRNA dysregulation in colorectal cancer: a clinical perspective. Br J Cancer. 2011;104(6):893-8.
- Ju J. miRNAs as biomarkers in colorectal cancer diagnosis and prognosis. Bioanalysis. 2010;2(5):901-6.
- Oh JS, Kim JJ, Byun JY, Kim IA. Lin28-let7 modulates radiosensitivity of human cancer cells with activation of K-Ras. Int J Radiat Oncol Biol Phys. 2010;76(1):5-8.
- Ragusa M, Statello L, Maugeri M, Majorana A, Barbagallo D, Salito L, et al. Specific alterations of the microRNA transcriptome and global network structure in colorectal cancer after treatment with MAPK/ERK inhibitors. J Mol Med (Berl). 2012;90(12):1421-38.
- Rupaimoole R, Han HD, Lopez-Berestein G, Sood AK. MicroRNA therapeutics: principles, expectations, and challenges. Chin J Cancer. 2011;30(6):368-70.
- Li C, Feng Y, Coukos G, Zhang L. Therapeutic microRNA strategies in human cancer. AAPS J. 2009;11(4):747-57.
- Christopher AF, Kaur RP, Kaur G, Kaur A, Gupta V, Bansal P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect Clin Res. 2016;7(2):68-74.
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487. 2012;487(7407):330-7.
- Fearon ER, Carethers JM. Molecular subtyping of colorectal cancer: time to explore both intertumoral and intratumoral heterogeneity to evaluate patient outcome. Gastroenterology. 2015;148(1):10-3.
- Pan JS, Hong MZ, Ren JL. Reactive oxygen species: a double-edged sword in oncogenesis. World J Gastroenterol. 2009;15(14):1702-7.
- Ogrunc M, Di Micco R, Liontos M, Bombardelli L, Mione M, Fumagalli M, et al. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014;21:998-1012.
- D' Abaco GM, Whitehead RH, Burgess AW. Synergy between Apc min and an activated ras mutation is sufficient to induce colon carcinomas. Mol Cell Biol. 1996;16(3):884-91.
- Moon BS, Jeong WJ, Park J, Kim TI, Min do S, Choi KY. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/β-catenin signaling. J Natl Cancer Inst. 2014;106(2):373.
- Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131(4):1096-109.
- Li J, Mizukami Y, Zhang X, Jo WS, Chung DC. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3beta. Gastroenterology. 2005;128(7):1907-18.
- Castellano E, Downward J. RAS Interaction with PI3K: More than Just Another Effector Pathway. Genes Cancer. 2011;2(3):261-74.
- Jeong WJ, Yoon J, Park JC, Lee SH, Lee SH, Kaduwal S et al. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci Signal. 2012;5(219):30.