Case Report
Lipofibromatosis of the Paravertebral Soft Tissue in a Child of Two Years Old
Colangeli S1*, Colangeli M2, Gasbarrini A3, Righi A4 and Boriani S3
1Department of Orthopaedic Oncology and Reconstructive Surgery, Careggi University Hospital, Florence, Italy
2Department of Musculo-Skeletal Oncology, Rizzoli Orthopaedic Institute, Bologna, Italy
3Department of Oncological and Degenerative Spine Surgery, Rizzoli Orthopaedic Institute, Bologna, Italy
4Surgical Pathology, Rizzoli Orthopaedic Institute, Bologna, Italy
*Corresponding author: Colangeli S, Department of Orthopaedic Oncology and Reconstructive Surgery Careggi University Hospital, Largo Piero Palagi, 1. 50134, Florence, Italy
Published: 14 Apr, 2017
Cite this article as: Colangeli S, Colangeli M, Gasbarrini A,
Righi A, Boriani S. Lipofibromatosis of
the Paravertebral Soft Tissue in a Child
of Two Years Old. Clin Oncol. 2017; 2:
1262.
Abstract
Lipofibromatosis is a bland fibroproliferative process that invades skeletal muscle. Typically the
children are affected before 8 years with a peak under 2 years old. Head, neck or thigh are the most
frequently involved sites.The management strategies are different: the first, periodical clinical and
imaging control with MRI to evaluate the symptoms and the size of the tumor mass; the second, to
assess the feasibility and to perform chemotherapy in selected cases; the third, surgical excision of
the tumor. This article reports on the youngest patient diagnosed with this rare soft tissue tumor in
thoracolumbar area. In fact, according to the Authors’ knowledge, no other cases of this tumor in
this localization have been presented in the literature. Our study demonstrated that surgical excision
is the gold treatment choice in this tumor when feasible. In fact, the scoliosis caused by tumor
stopped its evolution after its surgical excision.
Keywords: Lipofibromatosis; Spine; Tumors
Introduction
There are two types of lipofibromatosis: one type identical to adult desmoidfibromatosis
and the second like a diffuse mesenchymal type [1]. Both type often containing fat that usually involves head, neck or thigh. Alternate/Historical Names are: aggressive infantile fibromatosis, cellular fibromatosis, congenital fibromatosis, diffuse mesenchymal fibromatosis, fibrosarcoma-like
fibromatosis and infantile desmoid-type fibromatosis [2].
Resonance Imaging (MRI) usually shows a soft tissue lesion with high fat content [3,4].
Excision and histopathological analysis reveal a tumour composed of two patterns [3]: immature or diffuse pattern and uniform bland cells in myxoid stroma (nuclei round or oval to spindled and
scant cytoplasm).
There is no evidence for the effectiveness of chemotherapy in this disease and surgical excision
seems to be the only possible curative therapy even if recurrences are frequent following incomplete
excision [1,5].
The patient here reported was submitted to excisional surgery and resulted in excellent
conditions without any sign of recurrence at a follow-up of five years.
Case Report
A.F., male, 29 months old, Caucasian, was referred to our institution with a big painless mass
in the thoracolumbar area at right. At the age of 3 months, his parents noticed a “lump” in the
right part of the back. At 18.5 month a consultation was performed in another Institution, where
a standard radiogram (Figure 1a) showed a thoracolumbar scoliosis (16°Cobb angle with right concavity) and MRI (Figure 2a) described a soft tissue mass in the paraspinal zone from T7 to L4. An ultrasonography-guided biopsy was made at the age of 20 months; histopathologic diagnosis
was lipofibromatosis. After this diagnosis the patient underwent chemotherapy with Vinblastine
and methotrexate weekly during 7 months. Three gadolinium MRIs were performed to check
mass evolution, reporting that the tumour remained unchanged. At the referral to our Institution,
the pathologist confirmed to diagnosis of lipofibromatosis. The medical oncologist remarked that further chemotherapy had no supporting evidence. En Bloc
resection planned to achieve wide margins was performed at the age
of 32 months. The specimen size was 13cm x8cmx5.5 cm. Histology
showed an admixture of fibroblastic spindle cells and mature adipose
tissue (Figure 3a). Poorly demarcated lobules of fat were separated
and traversed by thickened fibroblastic septa. The septa comprised
spindle cells showing no atypia or pleomorphism, and minimal
amounts of collagen were present (Figure 3b). In addition there
were paucicellular fibrotic areas and myxoid change possibly due to
chemotherapy. At immunohistochemistry the neoplastic cells were
positive for vimentin and negative for smooth muscle act in, desmin,
actin muscle specific, caldesmon and beta-catenin. The proliferative
index Ki67 (clone MIB1) was 2%.
At five years post-operative follow-up no evidence of local disease
was found at clinical controls and also radiological examinations, performing during this period, like MRI (Figure 2b) and ultrasound
images resulted negative for local recurrence. The scoliosis remained
stable and of minor significance even at 5 years of follow-up
as demonstrated by the whole spine radiographs performed in
orthostatic position at this time that did’t show worsening by the first
(Figure 1b).
Figure 1a
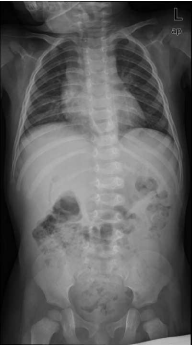
Figure 1a
First X-ray pre op Cobb 16° (18.5 months of life).
Figure 1b
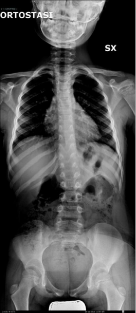
Figure 1b
Latest X-ray performed 5-years after surgery: Cobb angle
is conserved and scoliosis was not worsened in comparison with first
radiographs.
Figure 2a
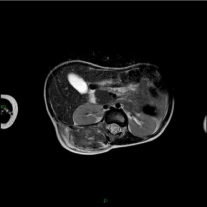
Figure 2a
First MRI preoperative, extension of the lesion in the soft tissues
of paravertebral muscles from T7 to L4.
Figure 2b
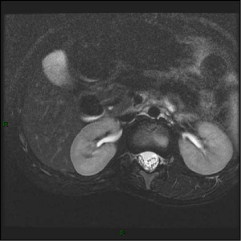
Figure 2b
Post-operative MRI performed 2 years after surgery didn’t show
local recurrence.
Discussion
Lipofibromatosis is an uncommon tumor typically found on the
distal extremities of infants, although it can appear in various sizes
and locations andit should be considered in the differential diagnosis
of pediatric soft tissue neoplasms [6]. Clinically, the masses are
painless and slow-growing [3] but they can cause deformity in the
antomical locations where they are present [7]. Surgical excision is
the the treatment of choice, while the role of chemotherapy is not
supported except in the cases of desmoid-type fibromatosis that is
recurrent or not amenable to treatment with surgery [8]. Small
number of cases with diagnosis of lipofibromatosis are reported in
literature because it is a rare entity.
Fretsch JF et al. [1] described lipofibromatosis for the first
time in 2000, as a rare pediatricneoplasm that has been variously
interpreted as a type of infantile or juvenile fibromatosis, a variant
of fibrous hamartoma of infancy, and a fibrosinglipoblastoma. This
variability of interpretations could explain the lack of previous
reports. Microscopic examination of 45 cases of soft tissue tumors
revealed abundant adipose tissue with a spindled fibroblastic element
that chiefly involved the septa of fat and skeletal muscle [9]; these
particular findings guide them to propose a new classification for
these lesions. In this series regrowth of the tumor or persistent disease was documented in 72% of the cases [1]. After this first
report, other 6 cases around were registered at present. Marzban
and Geramizadeh [10] reported in 2012 a case of lipofibromatosis
associated with syndactilia, bilateral complete cleft lip and palate,
trigonocephaly, and atrial septal. Herrmann et al. [11] described in
2004 a young girl with a neck mass described as Lipofibromatosis.
Friesenbichler and Leithner [12] published in 2010 a case in an adult
female of 25 years with a retroclavicular growing lipofibromatosis
that caused recurrent paresthesia. Kenney et al. [13] reported in
2007 a case of lipofibromatosis in a 5-year old boy which revealed an
apparently balanced three-way translocation, t(4:9:6).
Lipofibromatosis lacks the primitive mesenchymal islands with
myxoidstroma that characterize fibrous hamartoma of infancy;
discrimination from desmoid-type fibromatosis is based in large
part on the degree of preservation of larger fat lobules and the lack
of solid fibrous growth. Lipoblastoma can be distinguished based
on its constituent of immature adipocytes and the lack of a striking
spindle cell component [3,13]. Anomalies involving 6q23~q27 have
been implicated in a number of different and seemingly unrelated
neoplasms, however, including lymphoid malignancies [14],
malignant fibrous histiocytoma [15], angiofibroma [16], and lipomas
[17].
This paper reports the youngest patient diagnosed with this rare
soft tissue tumor in thoracolumbar area. No other cases with this
diagnosis and localization of this tumor are presented in literature.
The teaching point of this case is that surgical excision is the gold
standard in the treatment of this disease. In fact scoliosis presented
at the diagnosis and caused by the presence of lipofibromatosis in
the paravertebral soft tissues stopped its evolution after the surgical
excision of the tumor and this result was confirmed also at 5 years of
follow-up.
Figure 3a
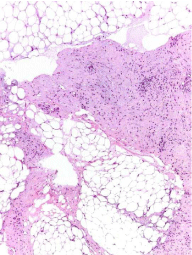
Figure 3a
Histological features: neoplasm constituted of an admixture of
fibroblastic spindle cells and mature adipose tissue (magnification 200X).
Figure 3b
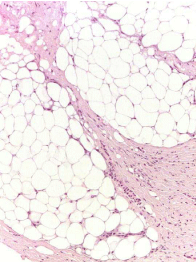
Figure 3b
Histological features: without cytological atypia or pleomorphism
(magnification 400X).
References
- Fetsch JF, Miettinen M, Laskin WB, Michal M, Enzinger FM. A clinicopathologic study of 45 pediatric soft tissue tumors with an admixture of adipose tissue and fibroblastic elements, and a proposal for classification as lipofibromatosis. Am J SurgPathol. 2000;24(11):1491-1500.
- Sargar KM, Sheybani EF, Shenoy A, Aranake-Chrisinger J, Khanna G. Pediatric Fibroblastic and Myofibroblastic Tumors: a Pictorial Review. Radiographics. 2016;36(4):1195-214.
- Vogel D, Righi A, Kreshak J, Dei Tos AP, Merlino B, Brunocilla E, et al. Lipofibromatosis: magnetic resonance imaging features and pathological correlation in three cases. Skeletal Radiol. 2014;43(5):633-9.
- Sheybani EF, Eutsler EP, Navarro OM. Fat-containing soft-tissues in children. Pediatr Radiol. 2016;46(13):1760-3.
- Samarakoon L, Fernanado T, Liyanage E, Wirithamulla H, Perera KS. Unusual case of recurrent thigh lump in a girl: a case report. J Med Case Rep. 2014;8:245.
- Boos MD, Chikwava KR, Dormans JP, Chauvin NA, Jen M. Lipofibromatosis: an institutional and literature review of an uncommon entity. Pediatr Dermatol. 2014;31(3):298-304.
- Joseph G, Zenios M. Congenital bowing of the tibia due to infantile lipofibromatosis corrected with a Taylor Spatial Frame. Musculoskelet Surg. 2014;98(3):247-50.
- Skapek SX, Ferguson WS, Granowetter L, Devidas M, Perez-Atayde AR, Dehner LP, et al. Vinblastine and methotrexate for desmoid fibromatosisin children: results of Pediatric Oncology Group Phase II Trial. J Clin Oncol. 2007 Feb 10;25(5):501-6.
- Fletcher CDM, Unni KK, Mertens F, editors in Pathology and genetics of tumours of soft tissue and bone. World Health Organization Classification of Tumours. Lyon. 2002. IARC Press.
- Marzban S, Geramizadeh B.Lipofibromatosis accompani ed by several congenital anomalies, report of a rare case. Indian J PatholMicrobiol. 2012;55(2):242-4.
- Herrman BW, Dener LP, Forsen JW.Lipofibromatosis presenting as a pediatric neck mass. Int J PediatrOtorhinolaryngol. 2004;68(12):1545-9.
- Friesenbichler J, Leithner A, Beham A, Windhager R.Retroclavicularlipofibromatosis: case report and review of the literature. EurOrthopTraumatol. 2010;1(3-4):115-118.
- Kenney B, Richkind KE, Friedlaender G, Zambrano E. Chromosomal rearrangements in lipofibromatosis. Cancer Genet Cytogenet. 2007;79(2):136-9.
- Cook JR, Shekhter-Levin S, Swerdlow SH. Utility of routine classical cytogenetic studies in the evaluation of suspected lymphomas: results of 279 consecutive lymph node/extranodal tissue biopsies. Am J ClinPathol. 2004;121:826-35.
- Simons A, Schepens M, Jeuken J, Sprenger S, van de Zande G, Bjerkehagen B, et al. Frequent loss of 9p21(p16INK4A) and other genomic imbalances in human malignant fibrous histiocytoma. Cancer Genet Cytogenet. 2000;118(2):89-98.
- Dietrich CU, Krone W, Hochsattel R. Cytogenetic studies in tuberous sclerosis. Cancer Genet Cytogenet. 1990; 45(2):161-177.
- Sreekantaiah C, Leong SP, Karakousis CP, McGee DL, Rappaport WD, Villar HV, et al. Cytogenetic profile of 109 lipomas. Cancer Res. 1991;51(1):422-33.