Mini Review
The Role of the Insulin Receptor Isoforms in the Insulin-Like Growth Factor Signaling Axis in Cancer
Brianne L Sanford1 and Dawn S Chandler1,2*
1Center for Childhood Cancer and Blood Diseases, The Research Institute at Nationwide Children’s Hospital, USA
2Department of Pediatrics, The Ohio State University, USA
*Corresponding author: Dawn S Chandler, Department of Pediatrics, The Ohio State University, USA
Published: 21 Mar, 2017
Cite this article as: Sanford BL, Chandler DS. The Role
of the Insulin Receptor Isoforms in the
Insulin-Like Growth Factor Signaling
Axis in Cancer. Clin Oncol. 2017; 2:
1236.
Abstract
The Insulin-like Growth Factor (IGF) signaling system is known for regulating critical cellular
processes related to growth and apoptosis. The IGF-axis is activated upon IGF stimulation of the
IGF-1 receptor (IGF-1R), and it has been well demonstrated that increased expression of IGF-1R and
the IGF-2 ligand is implicated in cell transformation and tumor propagation. For several decades,
efforts have been focused on developing treatment strategies aimed at inhibiting IGF-1R action but
produced suboptimal results. Recent research has confirmed the presence of an autocrine signaling
loop involving IGF-2 and an insulin receptor isoform A (IR-A), which is produced as a result of
alternative splicing of the IR. Stimulation of full-length IR (IR-B) by insulin typically activates
glucose regulating pathways, whereas IR-A activation by IGF-2 leads to mitogenic signaling. IR-A
is a naturally occurring isoform, prevalent during fetal development and in certain tissues. It is also
shown to be highly upregulated in many cancer types, increasing the cancer cell’s responsiveness
to IGF-2 thus providing the tumor cell a growth advantage. In this mini review, we discuss the
mechanism of IR alternative splicing and its role in cancer and treatment resistance.
Keywords: Cancer; IGF-2; Insulin receptor; Insulin-like growth factor-1 receptor; Splicing
Introduction
The Insulin-like Growth Factor (IGF) system plays an important role in the regulation of
cellular growth and development. Many cancers overexpress IGF hormones and receptors leading
to enhanced autocrine and paracrine signaling to promote growth and inhibit apoptosis, presenting
the tumor cell with a growth advantage [1-6]. The IGF system is therefore, an attractive target for
cancer therapeutic development. Recent therapeutic development has targeted the IGF-axis using
various antibody and small molecule approaches against the IGF-1 receptor (IGF-1R) and the related
Insulin Receptor (IR), as well as the ligands that activate these receptors. This approach to slow
tumor growth by inhibiting the IGF signaling system showed promise in preclinical development
but clinical trial results were disappointing [7]. Recently evaluated therapeutic strategies focused on
the IGF-signaling pathway have been reviewed previously [8].
It is well known that intracellular signaling pathways are very complex and involve a high level of
interconnectivity [9]. Crosstalk between the IGF-1R and IR signaling pathways is likely a key factor
in the failure of these clinical trials [10-13]. Insulin Receptor isoform A (IR-A) is stimulated by IGF-
2, which leads to activation of mitogenic signaling, bypassing the IGF-1R inhibitors. This IR isoform
is often upregulated in many cancers and has been the subject of recent interest since it is becoming
clear that therapeutic strategies should consider the IR signaling family. This review summarizes the
prevalence and mechanism of IR-A in cancer and its role in IGF-1R-targeted therapies.
The IGF and insulin signaling family
The IGF and insulin signaling pathways are activated by IGF-1, IGF-2 or the homologous
hormone insulin. These factors activate at least six receptors: IGF-1R, two forms of the IR produced
from alternative splicing (IR-A and IR-B), and various hybrid receptors (Figure 1). Both IGF-1R and
IR are transmembrane tyrosine kinase receptors that function as a dimer. Each monomer consists
of an extracellular α subunit and a membrane-spanning β subunit, both synthesized from a single
mRNA. The protein is cleaved by furin into the two subunits linked by disulfide bonds to form the
αβ chain, which dimerizes to form the functional receptor [14].
In addition to the homodimer receptors, αβ chains from IGF-1R and IR can dimerize with
each other to make heterodimers receptors referred to as hybrid receptors. To add another layer of complexity to the IGF and insulin signaling family, there are
two isoforms of the IR that result from alternative splicing of exon
11 during maturation of the pre-mRNA. The full-length receptor is
known as IR-B and includes exon 11, which resides at the C-terminus
of the α chain and is predicted to influence ligand binding. On the
other hand, IR-A lacks exon 11, which allows increased affinity for
signaling ligands in addition to insulin. This alternatively spliced exon
is only 36 nucleotides in length and encodes for 12 amino acids but
the receptor lacking exon 11 is able to bind IGF-2 with high affinity,
unlike the full-length IR-B receptor [15,16].
Splicing is influenced by proteins that bind specific sequences of
the pre-mRNA. These splicing factors can either recruit or block the
spliceosomal snRNPs, leading to either the recognition of specific
exons and removal of the intervening introns, or the silencing of
splicing signals at exon boundaries, resulting in the removal of
one or a series of exons and introns. The regulation of IR exon 11
splicing is controlled by repressor and enhancer sequences in exon
11 and the surrounding introns [17]. Positive regulators of splicing
that promote exon 11 inclusion include muscle blind-like splicing
regulator 1 [18-20] and serine/arginine-rich splicing factors 1 and
3 [21]. Alternatively, splicing factors that cause skipping of exon 11
include heterogeneous nuclear riboprotein A1 [22] and CUG-binding
protein 1 [21]. Expression of the IR isoforms is developmentally
regulated. Fetal tissue including brain, muscle, liver, kidney, and
fibroblasts were assayed for IR-A expression and compared to adult
tissues. In all fetal tissues except brain, there was higher expression
of IR-A than the respective adult tissues [15]. Expression of the two
IR isoforms is also tissue specific. The full-length form can be mostly
found in insulin-sensitive tissues such as liver, muscle, adipocytes and
kidney whereas the IR-A form is widely expressed [23]. Both forms
of the insulin receptor can produce homo- and heterodimers as well
as hybrid receptors with IGF-1R allowing for crosstalk between these
two receptor families and for a complex signaling axis [24].
Binding affinity and receptor activation for IGF-1R and both forms
of IR receptors has been characterized [15,25]. IGF-1R is activated by IGF-1 and IGF-2, which bind to the extracellular α-subunit of the
receptor and cause a conformational change in the β-subunit. This
leads to autophosphorylation of the β-subunit and recruitment of
adapter proteins and subsequent activation of mitogenic signaling
cascades including Mitogen Activated Protein Kinases (MAPK)
and phosphatidylinositol 3-kinase (PI3K)-Akt pathways to promote
cell growth and motility and antiapoptotic signaling [26-28].
Both forms of the IR have high affinity for insulin, which leads to
primarily metabolic effects through PI3K-Akt signaling. It is also
known that IGF-2 can stimulate the IR to activate the mitogenic
pathways. Previous studies using mouse fibroblasts deficient for
IGF-1R and expressing low levels of IR failed to proliferate in serumfree
conditions when stimulated with growth factors. When IR is
expressed, these cells are able to grow when stimulated with IGF-2
[29]. Subsequent studies demonstrated that IR-A is the isoform that
is stimulated by IGF-2, activating mitogenic pathways and allowing
for proliferation of these receptor-deficient fibroblast cell lines [15].
IR-A binds IGF-2 and is auto-phosphorylated with relatively
high affinity, whereas IR-B only has strong affinity for insulin. It
has also been shown that hybrid receptors with IGF-1R and IR-A
can also bind IGF-2 along with IGF-1 and insulin (Figure 1) [30].
Activation by these two types of ligands leads to activation of distinct
cellular processes. When the receptors bind IGFs or insulin, this
induces structural changes and subsequent auto-phosphorylated of
tyrosine residues like that of IGF-1R.While IR-B signaling promotes
mainly metabolic processes related to glucose homeostasis, IR-A also
activates mitogenic signaling cascades when stimulated by IGF-2.
IR-A and cancer
It is becoming increasingly evident in recent decades the
prevalence of IR-A in a variety of cancer cell types. A number of labs
have reported increased IR-A:IR-B ratios in a number of neoplasms
including breast, colon, lung, thyroid, liver, and bone cancers
[15,31-34]. It was reported by Sciacca and colleagues [15] that breast
cancer tissue samples had increased IR-A expression (40-80% IRA)
as compared to normal breast tissue (30-50% IR-A). They also
determined IGF-2 stimulated breast cancer cell growth. Moreover,
the potency of IGF-2 was correlated to IR-A expression, indicating
the presence of autocrine and paracrine signaling via the IGF-2/
IR-A interaction. Similar overexpression of IR-A in thyroid cancer
cells and tissue specimens was reported [32]. Interestingly, poorly
differentiated thyroid cells produced IGF-2 and overexpressed IR-A,
again indicating the presence of an autocrine loop promoting cancer
cell growth. Similar lines of investigation confirmed over expression
of IR-A in prostate, lung, leiomyosarcoma, osteosarcoma, and colon
cancer cells and tissue samples, further emphasizing the pervasiveness
of IGF-2/IR-A signaling in cancer cells [12,15,31,34-36].
IR-A resistance in IGF-1R therapies
Therapeutic strategies have focused on inhibiting tumor growth
through the IGF-1R signaling axis due to the frequent over expression
of this receptor in cancer cells and its key role in regulating cell
proliferation and apoptosis. The three most investigated strategies
include: receptor-targeting antibodies, tyrosine kinase inhibition, and
ligand-targeting neutralization antibodies [8]. Therapies targeted to
IGF-1R have been promising but there is evidence to suggest that the
IR compensates for IGF-1R inhibition as this single line of therapy
is not sufficient to inhibit tumor growth. A recent study of Ewing’s
sarcoma investigated the effect of several anti-IGF-1R therapies,
and the researchers found that cells not responding to these drugs had higher expression of IR-A. They report that tumors with a low
ratio of IGF-1R:IR are unlikely to benefit from anti-IGF-1R therapies
[37]. They also conclude Ewing’s sarcoma cells may adapt to anti-
IGF-1R therapies through activation of an IR-A-dependent pathway.
Increased IGF-2 expression was also noted in resistant cells indicating
a switch from IGF-1/IGF-1R to IGF-2/IR-A dependency to maintain
mitogenic signaling pathways.
A recent study by Forest and colleagues investigated the
correlation of resistance of cixutumumab, an anti-IGF-1R and
IR and IGF-1R expression [12]. By analyzing transcript levels
from tissue samples in their tumor models (Rhabdoid, Ewing’s
sarcoma, rhabdomyosarcoma, glioblastoma, neuroblastoma and
osteosarcoma), they discovered that high IR expression levels
indeed correlated to poor antitumor efficacy of cixutumumab. It is
interesting to note that IR-A was the predominant form present in
the tissue samples and IR-B expression was rather weak but IR-A
expression alone failed to correlate cixutumumab efficacy, suggesting
that both forms of IR may contribute to anti-IGF-1R resistance. In
experiments using stably induced breast cancer cells over expressing
IR-B, treated with cixutumumab or an anti-IGF-2 neutralizing drug,
there was only partial inhibition of colony formation. However, when
cells were treated with both therapies, the drug resistant phenotype
was reversed, suggesting that IGF-2 is implicated in the resistance
of tumor phenotypes by IR-B along with previously reported IR-A
resistance to anti-IGF-1R therapies [13]. It is also possible that
resistance is conferred through IR and IGF-1R hybrid receptors
but it has been shown that cixutumumab and other anti-IGF-1R
therapies are very effective at receptor internalization and subsequent
degradation, effectively neutralizing hybrid receptors [38].
Figure 1
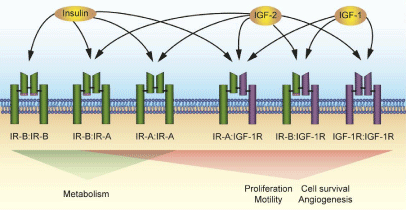
Figure 1
The insulin and IGF-1 receptor family.
The insulin and IGF-1 receptors exist as homodimers or heterodimers, each
with distinct ligand binding preferences and signaling outcomes. Each “half”
of the dimer is comprised of an αβ chain synthesized from one pre-mRNA
transcript that is proteolytically cleaved and joined by disulfide linkages
(black bars). IR (green receptor) is expressed as two isoforms, differing by
the presence or absence of exon 11 (pink box). Both the full-length IR-B
and exon 11 deficient IR-A isoforms dimerize to form functional receptors or
hybrid receptors as indicated below each receptor. IGF-1R (purple receptor)
can also form functional receptors with both IR isoforms. The IR-B has strong
affinity for insulin, which results in signaling of metabolic pathways. IGF-
1R activation by either IGF-1 or IGF-2 leads to processes promoting cell
proliferation and motility and antiapoptotic signaling. Hybrid receptors with
IR and IGF-1R as well as the homodimer IR-A activate mainly mitogenic
pathways when stimulated by IGF-2 [25,30].
Conclusion
IGF-1R is expressed at high levels in several tumor types inclusive of breast, ovarian, prostate, head and neck, and squamous lung cancer and tumor types [12]. The IGF-signaling axis plays a key role in promoting cellular proliferation and inhibiting apoptosis and represents an attractive and heavily developed avenue for therapeutic development. Another key player in this complex signaling pathway is the IR, which exists in two isoforms that result from alternative splicing and give rise to receptors with different ligand affinities and signaling outcomes. It is becoming apparent that the unexpected poor performance of therapies targeting the IGF-signaling axis through IGF-1R inhibition and ligand neutralization can be attributed to IGF-2 stimulation of IR-A, activating mitogenic pathways and circumventing this therapeutic strategy. A recent study highlighted the prevalence of IR-A expression with an RNA-seq analysis that evaluated RNA expression in 6,943 samples representing 21 tumor types and found IR-A to be present in all tumor types. This same study also provides evidence for IGF-2 stimulation through IR-B to promote cellular growth, further complicating this signaling cascade [12]. It is clear that co-targeting of IGF-1R and IR is necessary for an effective therapeutic strategy. This will represent a delicate task since IR expression is required for glucose metabolism, a critical cellular process. Therefore, it is critical to fully understand the action of the two IR isoforms and their role in mitogenic and metabolic signaling pathways to effectively target tumor cells and to develop ways to overcome tumor resistance associated with IR isoforms.
References
- Samani AA, Yakar S, LeRoith D, Brodt P. The Role of the IGF System in Cancer Growth and Metastasis: Overview and Recent Insights. Endocrine Reviews. 2007; 28: 20-47.
- Peters G, Gongoll S, Langner C, Mengel M, Piso P, Klempnauer J, et al. IGF-1R, IGF-1 and IGF-2 expression as potential prognostic and predictive markers in colorectal-cancer. Virchows Archiv. 2003; 443: 139-145.
- Moschos SJ, Mantzoros CS. The Role of the IGF System in Cancer: From Basic to Clinical Studies and Clinical Applications. Oncology. 2002; 63: 317-332.
- Shimizu C, Hasegawa T, Tani Y, Takahashi F, Takeuchi M, Watanabe T, et al. Expression of insulin-like growth factor 1 receptor in primary breast cancer: Immunohistochemical analysis. Human Pathology. 2004; 35: 1537-1542.
- Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A. 1993; 90: 11217-11221.
- Steller MA, Delgado CH, Bartels CJ, Woodworth CD, Zou Z. Overexpression of the Insulin-like Growth Factor-1 Receptor and Autocrine Stimulation in Human Cervical Cancer Cells. Cancer Research. 1996; 56: 1761-1765.
- Chen HX, Sharon E. IGF-1R as an anti-cancer target—trials and tribulations. Chin J Cancer. 2013; 32: 242-252.
- Singh P, Alex JM, Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Medical Oncology. 2013; 31: 805.
- Erin RK, Kwong-Kwok W. Insulin-like Growth Factor: Current Concepts and New Developments in Cancer Therapy. Recent Patents on Anti-Cancer Drug Discovery. 2012; 7: 14-30.
- Bid HK, Zhan J, Phelps DA, Kurmasheva RT, Houghton PJ. Potent Inhibition of Angiogenesis by the IGF-1 Receptor-Targeting Antibody SCH717454 Is Reversed by IGF-2. Molecular Cancer Therapeutics. 2012; 11: 649-659.
- Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999; 18: 2471-2479.
- Forest A, Amatulli M, Ludwig DL, Damoci CB, Wang Y, Burns CA, et al. Intrinsic Resistance to Cixutumumab is Conferred by Distinct Isoforms of the Insulin Receptor. Mol Cancer Res. 2015; 13: 1615-1626.
- Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci U S A. 2010; 107: 10791-10798.
- De Meyts P. The insulin receptor: a prototype for dimeric, allosteric membrane receptors? Trends in Biochemical Sciences. 2008; 33: 376-384.
- Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-Like Growth Factor II Receptor in Fetal and Cancer Cells. Mol Cell Biol. 1999; 19: 3278-3288.
- Denley A, Bonython ER, Booker GW, Cosgrove LJ, Forbes BE, Ward CW, et al. Structural Determinants for High-Affinity Binding of Insulin-Like Growth Factor II to Insulin Receptor (IR)-A, the Exon 11 Minus Isoform of the IR. Molecular Endocrinology. 2004; 18: 2502-2512.
- Kosaki A, Nelson J, Webster NJG. Identification of Intron and Exon Sequences Involved in Alternative Splicing of Insulin Receptor Pre-mRNA. J Biol Chem. 1998; 273: 10331-10337.
- Sen S, Talukdar I, Liu Y, Tam J, Reddy S, Webster NJG. Muscleblind-like 1 (Mbnl1) Promotes Insulin Receptor Exon 11 Inclusion via Binding to a Downstream Evolutionarily Conserved Intronic Enhancer. J Biol Chem. 2010; 285: 25426-25437.
- Echeverria GV, Cooper TA. Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Research. 2014; 42: 1893-1903.
- Grammatikakis I, Goo Y-H, Echeverria GV, Cooper TA. Identification of MBNL1 and MBNL3 domains required for splicing activation and repression. Nucleic Acids Research. 2011; 39: 2769-2780.
- Sen S, Talukdar I, Webster NJG. SRp20 and CUG-BP1 Modulate Insulin Receptor Exon 11 Alternative Splicing. Mol Cell Biol. 2009; 29: 871-880.
- Talukdar I, Sen S, Urbano R, Thompson J, Yates JR, Webster NJG. hnRNP A1 and hnRNP F Modulate the Alternative Splicing of Exon 11 of the Insulin Receptor Gene. PLoS ONE. 2011; 6: e27869.
- Seino S, Bell GI. Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun. 1989; 159: 312-316.
- Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, et al. Insulin and Insulin-like Growth Factor-I (IGF-I) Receptor Overexpression in Breast Cancers Leads to Insulin/IGF-I Hybrid Receptor Overexpression: Evidence for a Second Mechanism of IGF-I Signaling. Clin Cancer Res. 1999; 5:1935-1944.
- Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009; 30.
- Baserga R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene. 2000; 19: 5574-5581.
- Larsson O, Girnita A, Girnita L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br J Cancer. 2005; 92: 2097-2101.
- Sciacca L, Prisco M, Wu A, Belfiore A, Vigneri R, Baserga R. Signaling Differences from the A and B Isoforms of the Insulin Receptor (IR) in 32D Cells in the Presence or Absence of IR Substrate-1. Endocrinology. 2003; 144: 2650-2658.
- Morrione A, Valentinis B, Xu S-q, Yumet G, Louvi A, Efstratiadis A, et al. Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proc Natl Acad Sci U S A. 1997; 94: 3777-3782.
- Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/Insulin-like Growth Factor I Hybrid Receptors Have Different Biological Characteristics Depending on the Insulin Receptor Isoform Involved. J Biol Chem. 2002; 277: 39684-39695.
- Jiang L, Zhu W, Streicher K, Morehouse C, Brohawn P, Ge X, et al. Increased IR-A/IR-B ratio in non-small cell lung cancers associates with lower epithelial-mesenchymal transition signature and longer survival in squamous cell lung carcinoma. BMC Cancer. 2014; 14: 131.
- Vella V, Pandini G, Sciacca L, Mineo R, Vigneri R, Pezzino V, et al. A Novel Autocrine Loop Involving IGF-II and the Insulin Receptor Isoform-A Stimulates Growth of Thyroid Cancer. The J Clin Endocrinol Metab. 2002; 87: 245-254.
- Chettouh H, Fartoux L, Aoudjehane L, Wendum D, Clapéron A, Chrétien Y, et al. Mitogenic Insulin Receptor-A Is Overexpressed in Human Hepatocellular Carcinoma due to EGFR-Mediated Dysregulation of RNA Splicing Factors. Cancer Research. 2013; 73: 3974-3986.
- Avnet S, Sciacca L, Salerno M, Gancitano G, Cassarino MF, Longhi A, et al. Insulin Receptor Isoform A and Insulin-like Growth Factor II as Additional Treatment Targets in Human Osteosarcoma. Cancer Research. 2009; 69: 2443-2452.
- Perks CM, Zielinska HA, Wang J, Jarrett C, Frankow A, Ladomery MR. Insulin Receptor Isoform Variations in Prostate Cancer Cells. Frontiers in Endocrinology. 2016; 7.
- Sciacca L, Mineo R, Pandini G, Murabito A, Vigneri R, Belfiore A. In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene. 2002; 21: 8240-8250.
- Garofalo C, Manara MC, Nicoletti G, Marino MT, Lollini PL, Astolfi A, et al. Efficacy of and resistance to anti-IGF-1R therapies in Ewing's sarcoma is dependent on insulin receptor signaling. Oncogene. 2011; 30: 2730-2740.
- Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, et al. A Fully Human Monoclonal Antibody to the Insulin-Like Growth Factor I Receptor Blocks Ligand-Dependent Signaling and Inhibits Human Tumor Growth in vivo. Cancer Research. 2003; 63: 8912-8921.