Research Article
Pancreatic Alteration Induced by Incretins is Consistent with the Changes at the Early Stages of Pancreatic Carcinogenesis in the Hamster Model
Pour PM1,2* and Talmon G2
1Department of Pathology, UNMC Eppley Cancer Center, USA
2Department of Pathology and Microbiology, University of Nebraska, Medical Center, USA
*Corresponding author: Parviz M Pour, Department of Pathology, The UNMC Eppley Cancer Center, 600 S 42nd St, Omaha, NE 68198, USA
Published: 09 Sep, 2016
Cite this article as: Pour PM, Talmon G. Pancreatic
Alteration Induced by Incretins
is Consistent with the Changes
at the Early Stages of Pancreatic
Carcinogenesis in the Hamster Model.
Clin Oncol. 2016; 1: 1085.
Abstract
Background: GLP-1 analogs and DDP4 inhibitors, known as incretins, are used for the treatment of
type 2 diabetes. Although several published clinical and experimental studies point to the beneficial
effects of these drugs with negligible mild side effects, except for acute pancreatitis, the detailed
long-term effects of these drugs on the pancreas was missing. A recent report by the investigators at
UCLA showing a profound expansion of pancreatic endocrine cells, hyperplasia of ductal epithelium
and induction of endocrine lesions initiated serious concern about the safety of these drugs.
Methods: These alterations were almost identical to those found at the early stages of pancreatic
carcinogenesis in the hamster model. Therefore, in the present study, we compared the alterations
published by the UCLA investigators with the alterations of the pancreas occurring during
pancreatic carcinogenesis in the hamster model. Results and Discussion: The results heighten the
safety concern of these drugs and suggest that in cretins should be considered promoters of silent
malignant pancreatic lesions. The use of these drugs should be restricted to genuine long-standing
diabetics and be withheld from individuals with new-onset diabetes (Type 3 diabetes) who exhibit
asymptomatic pancreatic cancer.
Keywords: Glp-1; Pancreatic cancer; Islet
Introduction
Glucagon-like peptide 1 based therapies [GLP-1 receptor agonists (exenatide, liraglutide and
lixisenatide) and DPP-4 inhibitors (sitagliptin, vildagliptin, saxagliptin and linagliptin)] have been
available since 2005 for the treatment of Type 2 diabetes.
The efficacy of GLP-1 receptor agonists and DPP-4 inhibitors has been demonstrated. In terms
of safety, the most common adverse events seen in clinical trials with GLP-1 receptor agonists
are of gastrointestinal character, mainly nausea, vomiting and diarrhea. However, the incidence
diminishes over time. Other identified risks include pancreatitis, immunogenicity, acute renal failure
and rapid weight loss. Identified and potential risks with DPP-4 inhibitors include hypoglycemia,
hypersensitivity, gastrointestinal disorders, pancreatitis, skin disorders, transaminase elevation and
infections.
Although the manufacturers of the drugs have recently incorporated acute pancreatitis as a
serious side effect of the GLP-1 drugs, two fundamental concerns have been disregarded, including:
1. The long-term effects of the drugs on the structure of the pancreas and its consequences; and
2. The physiological and pathological effects of the drug and their duration after their cessation.
The first concern was highlighted by a study of Butler et al. [1].Using donor pancreases of Type 2
diabetics treated with in cretins, they demonstrated massive and extended proliferation of pancreatic
islet cells. Remarkably, the patterns of the morphological changes in their publication were almost
identical to the early lesions induced in the hamster pancreatic cancer model. The present report
compares the pancreatic lesions published by Butler et al. [1] with those found during pancreatic
carcinogenesis in the Syrian hamster model [2].
Material and Methods
Histological and immunohistochemical material from published article are presented [1].
In cretin Mimetics Products Liability Litigation is still on-going,
therefore, we were unable to obtain permission for the reproduction
of the photomicrograph published by Butler et al. The readers can
have access to the referenced figures by using the online version of
that publication with the following URL: http://www.ncbi.nlm.nih.
gov/pmc/articles/PMC3712065/
The presented figures from the hamster study (1974-2010) were
obtained from the Tumor Archive at the UNMC eppley Cancer
Center.
Figure 1
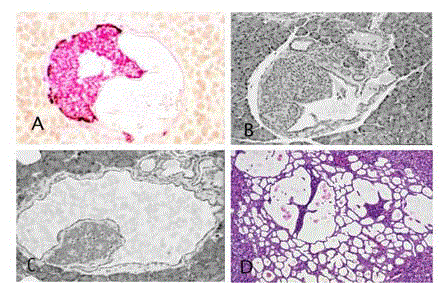
Figure 1
Intrainsular ductular patterns. A and B: Is lets of Syrian hamster
treated once with pancreatic carcinogen, N-nitrosobis (2-oxopropyl) amine
show cystic ducts (d)occupying almost half the islet(I) and are filled with
floccus material. Insulin, red and glucagon, brown. ABC method x 40. C and
D: Atrophic islets (I) attached to the wall of the cystic ducts directly (C) or by
a thin connective tissue (D). There are also numerous peri-insular ductular
structures. H&E x 40 (C), x20 (D). A,B and D are almost identical to Figure
2A published by Butler et al. [1].
Figure 2
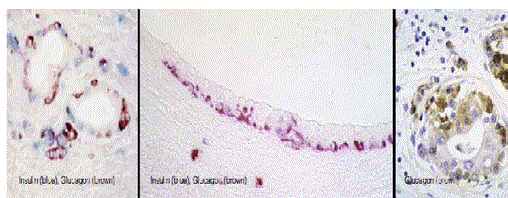
Figure 2
Large population of endocrine cells in malignant pancreatic
adenocarcinoma in humans (A and C) and hamsters (B). A: Insulin (blue),
glucagon (brown), somatostatin (black), ABC method, x40; B: insulin (red),
glucagon (brown), ABC, x40; C: insulin (brown), ABC, x 40. These figures are
strikingly similar to the figures C,D and E, respectively, published by Butler
et al. [1].
Results
As described earlier in detail in the hamster model [2, Chapter
12], the earliest alteration in carcinogen-treated animals is the
development of ductular structure within (intrainsular ducts) and
around the islets (peninsular duct). The initially tiny intrainsular
ductules gradually enlarge. Due to serous and mucinous secretion
by ductular cells, the ductules undergo cystic distention and the
increasing pressure of liquid within the intrainsular ductules causes
atrophy of the islet cells that appear as a small ortiny cell aggregate
attached to the cystic wall (Figure 1 and 2).In animals exposed to a
high dose of carcinogen the intra-and peri-insular ductules undergo
gradual hyperplasia, dysplasia and culminate in the malignant gland
that finally destroys the islets and invades the surrounding tissue
[2, pp 92-94]. In hamsters exposed to a single injection or repeated
low doses of the carcinogen, the earliest and the solitary alteration is
the formation of intrainsular ductules that either remain stationary
during the animal’s lifespan or slowly and gradually progress to
neoplastic lesions. Figure 2A in the publication of Butler et al.
[http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3712065/]
presents an identical pattern. In addition to the scattered islets of
various sizes in the figure, there is one large islet in the lower middle
field that contains a cystic duct and, in the upper middle field, there is
another cystic duct with an atrophic islet inside. The remaining three
cystic ducts appear to have lost the insular mass but still contain islet
cells within their epithelium.
Another striking feature in the induced lesions in the hamster
model, as well as in human pancreatic cancer, is the presence of
endocrine cells within the hyperplastic ducts, the number of which
may seem to exceed the number of the ductal cells (Figure 3).
Identical patterns have been observed in the study of Butler et al.
[1] shown in Figure 2C,D, and E. Mucinous ductal cell hyperplasia
bearing endocrine cells at the base of the epithelium, a characteristic
finding in human pancreatic cancer and in the hamster model has
also been demonstrated in Figure 7A and C of the publication(www.
ncbi.nlm.nih.gov/pmc/articles/PMC3712065/).
Figure 3
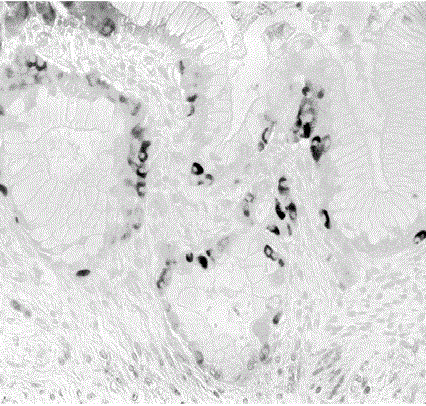
Figure 3
Human pancreatic mucinous adenocarcinoma with numerous
endocrine cells (black) in the basal epithelial layer. Identical lesions were
illustrated as figure 7A and 7C by Butler et al. [1] in diabetic patients treated
with incretin. ABC method, x 40.
Discussion
The present comparative study provides evidence that GLP-
1 treatment in type 2 diabetics causes profound alterations of the
pancreas, consistent with early lesions occurring during pancreatic
carcinogenesis in hamsters.
The origin of pancreatic cancer has remained controversial.
Genetic studies have suggested that under diverse experimental
conditions different pancreatic epithelial cells, including preexisting
acinar cells, pre-existing β-cells, pancreatic ductal cells,
and cells expressing the mesenchymal marker nestin, may undergo
malignant transformation. [3-8]. The development of the hamster
pancreatic cancer model, that in clinical, morphological, biological
and genetic aspects mimics the human disease [9], to the surprise
of many investigators, showed that islet cells play a major role in
pancreatic cancer development [2]. The initial lesion in hamsters is
the appearance of ductular structures within the islets in the form
of tiny conduits that gradually increase in size and undergo cystic or
malignant changes that totally destroy the islets. The alterations of
large ducts occur later [2, pp92-94]. The occurrence of intrainsular
ductules was thought to be related to the transdifferentiation of islet
cells to ductular cells, a process that was confirmed by several studies
[10-15]. The purified human and hamster pancreatic islets in culture
were shown to form ductal, acinar and intermediary cells (cells
with both exocrine and endocrine phenotypes) [10-15]. Hamster
islet cells treated in vitro with pancreatic carcinogen, N-nitrosobis
(2-oxopropyl) amine formed ductal adenocarcinoma in vivo [11].
Based on the silent clinical course of pancreatic cancer and difficulties
in dissecting the entire pancreas in serial sectioning, the possibility
that human pancreatic cancer also initiates from the islets remained
obscure.
The fundamental role of islet cells in pancreatic carcinogenesis
was evident by the observation that any procedure that stimulated
islet cell replication, such as induction of nesidioblastosis, feeding
a diet high in fat, which enlarges the islets, enhanced the cancer
formation [2, Chapter 17].Although the increased incidences of
pancreatic cancer in obese people, who have enlarged pancreatic cells
[16-19], appear to be in line with the findings in hamsters, the link
was bleak. The study by Butler et al. offered the missing link [1].
Some academic and industrial researchers [20,21] criticized
the paper of Butler et al. [1]. Although some of their points from
toxicological standards were reasonable, considering the parameters
that they have recommended, thousands or even more braindead
organ donors would have been required to obtain statistically
significant data. These authors did not propose how to obtain the
adequate number of treated and untreated sex-, age- and BMImatched
donor pancreases receiving the same agent (of more than
20 that are currently in use) and disregarded the simple fact that the
described unique alterations have never been reported in the pancreas
of any normal or diabetic persons treated or untreated with various
agents. Yet, the lesions were found only in the eight treated patients
but in none of the 14 controls [1].
The Committee for Medicinal Products for Human Use (CHMP)
reviewed the publication by an ad-hoc expert meeting held on July
10, 2013 and concluded that the results of the study by Butler et al.
[1] are not considered to constitute a new safety signal for the GLP-1
based therapies with respect to pancreatic safety. It was argued that,
due to the mechanism of action, there are still some uncertainties
with respect to the long-term pancreatic safety associated with these
products and updates to the risk management plans (including
planned and ongoing studies) and that harmonization of warnings in
the product information should be taken forward.
According to the assessment report for GLP-1 based therapies
25July 2013 EMA/474117/2013, in clinical trials, a few cases have been
reported for some products (Table 1). Although the data currently
available from clinical trials do not indicate an increased risk for
pancreatic cancer with these medicines, cases of pancreatic cancer
have been reported in the post-marketing setting. A cumulative
review of the cases has been undertaken and the majority (19 out of
29) had a time to onset of less than six months, a period considered
too short to suggest a causal relationship.
The role of GLP-1 analogs in pancreatic cancer varies widely
between the studies and based on the findings, multi-district litigation
(MDL) were established. Some trials, including SAVOR-TIMI (of
saxagliptin) and EXAMINE (of alogliptin), found no difference
between dipeptidyl peptidase 4 (DPP-4) inhibitor treatment and
placebo with regard to pancreatitis or pancreatic cancer [22]. In a
recent study, the risk was actually elevated for insulin. The pancreatic
cancer rate was 9.39% vs. 2.61% for population controls, with an
adjusted odds ratio of 3:6, suggesting that these drugs are strongly
associated with pancreatic cancer [22].
Although certain classes of nitrosamines act as pancreatic
carcinogenin hamsters [2, Chapter 7], the over production of insulin
seems to be the underlying factor in humans. Insulin is known as
a strong mitogenic factor and accelerates cell growth [23,24]. In
diabetics, where the insulin-producing beta cells have lost the ability
to produce insulin, GLP-1-enforced beta cell neogenesis and insulin
synthesis could well lead to the formation of immature beta cells or to
transdifferentiation of emerging beta cells to ductular cells associated
with errors in DNA synthesis and genetic mutations. The ability of
human islet cells to readily transdifferentiate into ductal (and acinar)
cells has been covered previously.
The fate of the lesions described [1] in the patients cannot be
predicted. Will it remain stationary, regress or progress to frank
malignancy? In hamsters, the described alterations occur following
low doses of pancreatic carcinogens and remain mainly stationary.
In some instances, the alterations advance to hyperplastic change,
indicating that the lesions virtually present the early stages of cancer
in these patients. It is possible that the advanced lesions have escaped
their detection. Although the observed morphological alterations
in the patients do not provide definite signs of malignancy and,
regretfully, the genetic alteration of the lesions was not performed,
the findings remain a controversial issue. In diabetics the amount of
locally secreted insulin and IGF-1 may not be sufficient to stimulate
the growth of the altered cells, but the GLP-1-induced insulin
certainly can affect the latent premalignant and small cancers, which
are reported to occur in up to 36% of individuals over 50years old
[25-37], and in a 9.5% incidence of “silent” pancreatic cancer” in
smokers [31]. These “silent” cancers are vulnerable to rapid growth
in any condition leading to increased insulin and IGF-1 production.
Support for this view is the increased pancreatic weight and ductal/
ductular hyperplasia in incretin-treated animals and patients [1]. The
reported and registered short latency of pancreatic cancer in clinical
trials, mentioned previously is in-line with this likelihood.
The problem with this assessment is the lack of data from
the pancreas of incretin-treated diabetics, the same as Butler and
associates. The ideal study to answer the question definitively would
involve large-scale pathohistological examination, databases from
multiple countries, providing enough data on new users as well as
prevalent users in order to eliminate bias from the duration of use.
Ideally, such studies would also last longer than any performed thus
far. Most cancer epidemiologists would like to see a drug exposure of
at least 8 to 10 years before they consider it usable.
Hence, the presented data suggest that incretins could act either
as promoters or the initiator of pancreatic cancer and their use should
be restricted to genuine long-standing diabetics and be withheld from
individuals with new-onset diabetes (Type 3 diabetes) who present
with asymptomatic pancreatic cancer [38-43].
Given the >20 million known patients with type 2 diabetes in the
United States alone, and the numerous GLP-1−based drugs either
available now or in the final stages of development, the potential
impact of the adverse effects of this class of drugs is considerable.
In summary, pushing the organism to perform a desirable function
(to produce insulin) that the body, for its own justified reasons, does
not want to do or is unable to do, can lead to unexpected, unwanted
and sometimes disastrous results.
Table 1
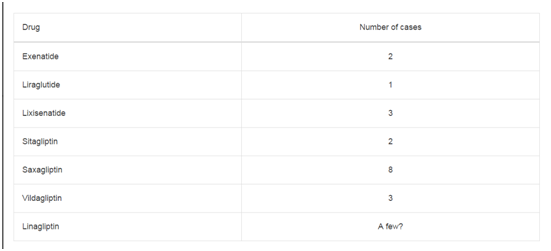
Table 1
In 9 out of 15 cases pancreatic cancer has been occurred to occur
within three months. In Saxagliptin group the time was 4-18 months. In others no
definite time lapse was recorded.
References
- Butler AE, Campbell-Thompson M, Gurlo T, Dawson D, Atkinson M, Butler P. Marked Expansion of Exocrine and Endocrine Pancreas with Incretin Therapy in Humans with Increased Exocrine Pancreas Dysplasia and the Potential for Glucagon-Producing Neuroendocrine Tumors. Diabetes. 2013; 62: 2595-2604.
- Pour PM. Hamster as a pancreatic cancer model.1984; 81-86.
- Irene Esposito, BjörnKonukiewitz, Anna Melissa Schlitter, Günter Klöppel. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. World J Gastroenterol. 2014; 20: 13833–13841.
- Maitra A, Leach SD. Disputed paternity: the uncertain ancestry of pancreatic ductal neoplasia. Cancer Cell. 2012; 22: 701–703.
- Ray KC, Bell KM, Yan J, Gu G, Chung CH, Washington MK. Epithelial Tissues Have Varying Degrees of Susceptibility to Kras G12D-Initiated. PLoS ONE. 2011; 6: e16786.
- Kong B, Michalski CW, Erkan M. From tissue turnover to the cell of origin for pancreatic cancer. Nature reviews Gastroenterology & hepatology. 2011.
- Kopinke LMD. Pancreatic Stem Cells. StemBook. 2008: 1–16.
- Ku HT. pancreatic progenitor cells--recent studies. Endocrinology. 2008; 149: 4312–4316.
- Pour PM. Why the hamster pancreatic cancer model is still the most useful tool for clinical studies. Journal of the Pancreas. 2013.
- Schmied BM, Ulrich A, Matsuzaki H, Ding X, Ricordi C, Moyer MP, et al. Maintenance of human islets in long term culture. Differentiation. 2000; 66: 173-180.
- Schmied BM, Ulrich A, Matsuzaki H, Li C-H, Pour PM. In vitro pancreatic carcinogenesis. AnnOncol. 1999; 10: 41-45.
- Pour PM, Schmied BM, Ulrich AB, Friess H, Andren-Sandberg A, Büchler MW. Abnormal differentiation of islet cells in pancreatic cancer. Pancreatology. 2001; 1: 110-116.
- Lucas-Clerc C, Massart C, Campion JP, Launois B, Nicol M. Long-term culture of human pancreatic islets in an extracellular matrix: morphological and metabolic effects. Mol Cell Endocrinol. 1993; 94: 9-20.
- Yuan S, Rosenberg L, Paraskevas S, Agapitos D, Duguid WP. Transdifferentiation of human islets to pancreatic ductal cells in collagen matrix culture. Differentiation. 1996; 61: 67-75.
- Kerr-Conte J, Pattou F, Lecomte-Houcke M, Xia Y, Boilly B, Proye C, et al. Ductal cyst formation in collagen-embedded adult human islet preparations. A means to the reproduction of nesidioblastosis in vitro. Diabetes. 1996; 45: 1108-1114.
- Naeye RL, Roode P. The sizes and numbers of cells in visceral organs in human obesity. Am J Clin Pathol. 1970; 54: 251-253.
- Ogilvie RF. The islands of Langerhans in 19 cases of obesity. J Pathol Bacteriol. 1993; 37: 473-481.
- Ogilvie RF. The endocrine pancreas in human diabetes. In: Brolin SE, Hellman B, Knutson (eds) The structure and metabolism of the pancreatic islet. Wenner-Gren Center International Symposium series 3. Pergamon Press. Oxford. 1964; 499-511.
- Butler AE, Galasso R, Matveyenko, Rizza RA, Dry S, Butler PC. Pancreatic duct replication is increased with obesity and tye 2 diabetes in humans. Diabetologia. 2010; 53: 21-26.
- Bonner-Weir S, In’t Veld PA, Weir GC. Reanalysis of study of pancreatic effects of incretin therapy: methodological deficiencies. Diabetes, Obesity and Metabolism. 2014: 16; 661-666.
- Harja E, Lord J, Skyler JS. An Analysis of Characteristics of Subjects Examined for Incretin Effects on Pancreatic Pathology. Diabetes Technology & Therapeutics. 2013; 15: 609-618.
- Miriam E Tucker. Study Suggests Incretin Drugs Don't Cause Pancreatic Cancer.
- vonWichert G, Jehle PM, Hoeflich A, Koschnick S, Dralle H, Wolf E, et al. nsulin-like growth factor-I is an autocrine regulator of chromogranin A secretion and growth in human neuroendocrine tumor cells. Cancer Res. 2000; 15: 4573-4581.
- Ohmura E, Okada M, Onoda N, Kamiya Y, Murakami H, Tsushima T, et al. Insulin-like Growth Factor I and Transforming Growth Factor α as Autocrine Growth Factors in Human Pancreatic Cancer Cell Growth Cancer Res. 1990; 50: 103-107.
- Kimura W, Morikane K, Esaki Y, Chan WC, Pour PM. Histological and biological patterns of microscopic ductal adenocarcinomas detected incidentally at autopsy. Cancer. 1998; 82: 1839-1849.
- Tomioka T, Andrén-Sandberg A, Fujii H, Egami H, Takiyama Y, Pour PM. Comparative histopathological findings in the pancreas of cigarette smokers and non-smokers. Cancer Lett. 1990; 55: 121-128.
- Kimura W, Morikane K, Esaki Y, Chan WC, Pour PM. Histological and biological patterns of microscopic ductal adenocarcinomas detected incidentally at autopsy. Cancer. 1998; 82:1839-1849.
- Sommers SC, Murphy SA, Warren S. Pancreatic duct hyperplasia and cancer. Gastroenterology. 1954; 27: 629.
- Collins JJ, Craighead JE, Brooks JR. Rationale for total pancreatectomy for carcinoma of the pancreatic head. N Engl J Med. 1966; 274: 599.
- Wilson C, Imrie CW. Occult pancreatic cancer with recurrent acute pancreatitis. Postgrad Med J. 1986; 62: 765-7675.
- Tomita T, Vacha E, Rengachery S, Watanabe I. Occult adenocarcinoma of the pancreas in a patient with Lindau’s disease. Am J Dig Dis. 1978; 23: 80–83.
- Compagno, Oertel E. Microcystic adenomas of the pancreas. A clinicopathologic study of 34 cases. A J C. 1978; 69: 289-298.
- Warshaw A, Compton C, Lewandrowski K, Carde- nosa G, Mueller P. Cystic tumors of the pancreas. Ann Surg. 1990; 212: 432-445.
- Fernández-del Castillo C, Targarona, J, Thayer SP, Rattner DW, Brugge WR, Warshaw AL. Incidental pancreatic cysts: Clinicopathologic Characteristics and Comparison With Symptomatic Patients. Arch Surg. 2003; 138: 427-434.
- Ariyama J, Suyama M, Ogawa K, Ikari T, Nagaiwa J, Fujii D, et al. The detection and prognosis of small pancreatic carcinoma. Int J Pancreatol. 1990; 7: 37-47.
- Manabe T, Miyashita T, Ohshio G, Nonaka A, Suzuki T, Endo K, et al. Small carcinoma of the pancreas. Cancer. 1988; 62: 135-141.
- Sataka K, Chung Y-S, Umeyama K, Takeuchi T, Kim YS. The possibility of diagnosing small pancreatic cancer (less than 4.0 cm) by measuring various tumor markers. Cancer. 1991; 68: 149-152.
- Permert J, Ihse I, Jorfeldt L. Pancreatic cancer is associated with impaired glucose metabolism. Eur J Surg. 1993; 159: 101–107.
- Schwarts SS, Zeidler A, Moossa AR, Kuku SF, Rubenstein AH. A prospective study of glucose tolerance, insulin, C-peptide, and glucagon responses in patients with pancreatic carcinoma. Am J Dig Dis. 1978; 23: 1107-1114.
- Gullo L, Pezzilli R, Morselli-Labate AM. Diabetes and the risk of pancreatic cancer. Italian Pancreatic Cancer Study Group. N Engl J Med. 1994; 331: 81-84.
- Karmody AJ, Kyle J. The association between carcinoma of the pancreas and diabetes mellitus. Br J Surg. 1969; 56: 362-364.
- Gullo L. Diabetes and the risk of pancreatic cancer. Ann Oncol. 1999; 4: 79-81.
- Cetin M, Colak R, Bayram F, Altinbas M, Unal A, Kelestimur F. High prevalence of diabetes in patients with pancreatic cancer in central Anatolia, Turkey. Diabetes Res ClinPract. 2002; 58: 97-100.