Research Article
Prognostic Factors for Ependymoma Survival: A Retrospective Study
Nizamutdinov D1,2#, Dandashi JA2#, Stock EM4, Kirmani BF1,2, Stroberg E3, Wu E1, Dayawansa S1, Huang JH1,2 and Fonkem E1,2,5*
1Department of Neurosurgery, Baylor Scott & White Health, USA
2College of Medicine, Texas A&M Health Science Center, USA
3Department of Pathology, Baylor Scott & White Health, USA
4Cooperative Studies Program Coordinating Center (CSPCC), VA Maryland Health Care System, USA
5Department of Neurology, Baylor Scott & White Health, USA
#These authors contributed equally to this work
*Corresponding author: Ekokobe Fonkem, Department of Neurology, Baylor Scott & White Health, Temple, Texas, 76504, USA
Published: 30 Aug, 2016
Cite this article as: Nizamutdinov D, Dandashi JA, Stock
EM, Kirmani BF, Stroberg E, Wu E, et
al. Prognostic Factors for Ependymoma
Survival: A Retrospective Study. Clin
Oncol. 2016; 1: 1063.
Abstract
Background: Ependymoma is a rare primary brain tumor that arises from the ependymal cells of
the intra-ventricular central nervous system.
Methods: Thirty-two clinical cases of ependymoma were obtained from the tumor registry of the
Scott & White Integrated Healthcare System from 1976 to 2013. We investigated the effects of
gender, age, race, tumor grade, surgical method, recurrence, radiation therapy (RT), chemotherapy
(CT), and mortality of patients.
Results: Fifty percent of patients had RT and 12.5% had CT. Tumor recurrence was observed in only
4 (12.5%) cases and all were diagnosed with grade II tumors. Sixteen patients (50.0%) underwent
subtotal resection, 11 (34.4%) gross total, and 5 (15.6%) underwent no surgical procedures. Twentytwo
patients (68.8%) are still living and 10 (31.3%) were deceased at time of analysis. Forty percent
of deceased were under 18 year of age. The median overall survival time for all patients was 15.2
years (182.5 months), with a 5-year survival rate of 80.0%. Patients with primary tumor sites in the
brain stem, frontal, and parietal lobes had survival rates of 87.5%, 100%, and 100%, respectively,
with no reported tumor recurrence (0.0% each).
Conclusion: Surgical treatment with attempted gross total resection was the most successful method
of ependymoma treatment. Primary tumor site is another important prognostic value for evaluation
of short and long-term outcomes of ependymoma diagnosed patients.
Impact: This study aims to identify novel prognostic factors for survival and to describe effective
treatments and outcomes of ependymoma diagnosed patients.
Keywords: Ependymoma; Prognostic factors; Survival; Outcomes; Epidemiology
Introduction
Ependymoma is a primary brain tumor that arises from the ependymal cells of the intraventricular
central nervous system (CNS) parenchyma. It is distinguishable from other CNS neoplasms by its
rarity and its propensity to afflict children [1].
Ependymomas account for 6-10% of pediatric intracranial tumors and represent 2-6% of
adult intracranial tumors [2-7]. Among pediatric cohorts, supratentorial ependymomas are
more common than infratentorial tumors and make up one-third of all ependymomas, whereas
infratentorial tumors are more common in adults [5,8-11]. Currently, the 5-year survival rates from
time of diagnosis for adults and children with ependymoma are 55-90% and 40-65%, respectively
[3,5,6,12,13]. Several studies have analyzed survival rates of patients less than or equal to 3 years
of age and these findings demonstrate that young children often have less favorable prognoses
than adult or older pediatric patients [9,13]. This disparity can most likely be attributed to the
fact that young children are often diagnosed when their disease has already progressed to a more
advanced stage, thereby imparting a less favorable prognosis as compared to a patient whose
ailment had been promptly observed and treated [4]. The World Health Organization (WHO)
data classify ependymomas into 3 groups by histopathology: grade
I (subependymoma or myxopapillary ependymoma), grade II
(classic ependymoma with cellular, papillary, clear cell, and tanycytic
variants), and grade III (anaplastic) [14]. Ependymomas of the
anaplastic variety are the most aggressive [15,16]. Some studies
have concluded that prospective research should be continued on
ependymomas before the anaplastic and classic varieties are more
clearly delineated [13,15,16]. However, patients presenting with
subependymomas or classic ependymomas can still display bleak
prognoses before treatment. With respect to subependymomas,
the size and location of the tumor is most helpful in determining
prognosis [2-5,17-21]. Few instances of extracranial metastases have
been recorded, but intraventricular metastases are relatively common
with grade II and grade III tumor variants. Although the current gold
standard of care for ependymoma treatment is gross total resection
(GTR) with concurrent radiation therapy (RT), studies have been
done comparing the relative efficacy of GTR against subtotal resection
(STR) in children and adults, depending on the location of their
ependymomas (infratentorial vs. supratentorial) [2,3,5,12,13,15-
17,22,23]. The most recurrences following treatment are local, but
RT decreases the probability of recurrence. The existence of relatively
few ependymoma cases precludes the ability to reach any unassailable
conclusions concerning appropriate treatment regimens. However,
most physicians agree that chemotherapy is relatively ineffective in
enhancing progression-free survival (PFS) for ependymoma patients
[8,13,16,24-27].
This study aims to evaluate the prognostic value of various
factors which could be utilized to generate more accurate predictions
regarding patient survival in individuals diagnosed with ependymoma,
including short and long-term outcomes. Further studies will be
needed to elucidate detailed analyses of tumor locations in different
regions of the brain with associated prognostic values of disease.
Materials and Methods
Sources of data and study population
All human investigations were performed after approval by an
institutional review board and in accordance with an assurance filed
with and approved by the US Department of Health and Human
Services.
Thirty-two total clinical cases of ependymoma were obtained from
the tumor registry of the Scott & White Integrated Healthcare System
from 1976 to 2013. There were no exclusion criteria and all cases
diagnosed with ependymoma were selected for this study. Age was
categorized into two groups: children (less than or equal to 18 years)
and adults (over 18). Race was categorized as white, Hispanic, black
non-Hispanic, and other/unknown, with white vs. non-white also
examined. Our categorization of ependymoma tumor grade fell into
3 groups as determined by histopathological studies: grade I, grade II,
and grade III. Data describing surgical method, RT, chemotherapy
(CT), tumor recurrence, and time to follow-up were also included
in data analyses. Surgical methods were described as STR, GTR, or
no surgical procedure (NSP). RT and CT were described as either
administered or not administered.
Data analysis
The data were incorporated from an Excel file into SAS, v9.2 (Cary,
NC), and R, v2.15.1 (The R Foundation for Statistical Computing) to
be analyzed for a number of variables. Descriptive statistics, including
frequencies and percentages, were calculated to describe patient
characteristics, tumor location, and mortality among ependymoma
cases. Comparisons for mortality among locations were examined,
overall and pairwise, using two-sample proportion tests. A type I
error of α=0.10 was assumed throughout given the smaller size of 32
for the sample. Kaplan-Meier curves were drawn for overall statistics
by gender, pediatric vs. adult, and white vs. other comparisons. Logrank
tests were used to compare mortality across groups. Quartile
estimates of median survival time were carried out with difficulty due
to the small sample sizes in use. Several of those median survival times
lacked a lower or an upper 95% Confidence Interval (CI) as a result.
Table 1
Table 1
Overall demographic characteristics of ependymoma patients (N=32).
Table shows descriptive values of overall occurrence of ependymoma by age,
race, gender, treatment modalities, tumor grades, recurrence of disease, median
overall survival time and 5 years survival rates, including time to follow up.
Results
Twenty-one (65.6%) patients were adults, and the remaining
11 (34.4%) were under or equal to 18 years of age. Twenty-three
patients (71.9%) were white, 5 (15.6%) Hispanic, 2 (6.3%) black
and 2 (6.3%) other/unknown ethnicity. The majority of patients
were male (59.4%). Three patients (9.4%) presented with grade III
anaplastic ependymoma, 21 (65.6%) with grade II ependymoma,
and the remaining 8 (25.0%) had grade I subependymoma. Sixteen
patients (50.0%) underwent STR, 11 (34.4%) GTR, and 5 (15.6%)
underwent NSP (Table 1). Among total 10 deceased patients, 8
(80.0%) underwent STR rather than GTR. RT was administered in
50% of all cases and CT in only 12.5%.
Eight patients (25.0% overall) became disease-free with no
recurrence at the time of analysis. Two of them (25.0%) were
treated only surgically. The remaining 6 (75.0%) were treated with
a combination of surgery and RT and/or CT. Among them, 5 were
administered RT and 1 had a combination of RT and CT. These
efforts resulted in 5 being alive (62.5%) at time of analysis.
Nine patients (28.1%) overall did not have remission of disease
after treatment. Seven surviving patients in this group received
surgical treatments. Four of them underwent surgery alone. Two
were treated with RT and CT, and one received RT in addition to
surgical resection. Two deceased patients (22.2%) underwent surgery
and CT alone, respectively.
Tumor recurrence was reported in only 4 cases (12.5% overall),
and all were grade II tumors, with 1 (25.0%) deceased. Data on tumor
recurrence were unavailable for 11 patients. Among patients with
unknown recurrence, 7 patients were diagnosed with grade II tumors,
3 with grade I tumors, and 1 with grade III tumor. Four (36.4%) were
deceased by the time of data analysis.
Forty percent of deceased patients were under the age of 18. Our
pediatric 5-year survival rate was 65.6% (95% CI 40.2-100%). The
adult 5-year survival rate was 78.4% (95% CI 57.9-100%). The median
overall survival time for 32 reported cases was 15.2 years (182.5
months), and the 5-year survival rate was 80.0%. The median time
to follow-up for those patients who are still living was 60.2 months,
and the median time to follow-up for the deceased patients was 74.6
months (Table 1).
Mortality by tumor location
We compared the incidence rates of the primary tumor sites
between the total ependymoma diagnosed population, the subsistent
treated, and deceased population to elucidate possible survival trends.
A side-by-side comparison is shown in Figure 1. These data suggest a
survival rate of 87.5% observed with primary tumor sites located in the
brain stem versus 62.5% elsewhere (p=0.186; Table 2). Survival with
tumors located in the frontal lobe was 100% versus 65.5% in other
locations (p= 0.220), with 100% survival for parietal lobe (p=0.325).
Among patients who died, accounting for 31.3% of all cases
(Table 2), and the most prevalent location was brain, NOS (40.0%),
followed by ventricle, NOS (30.0%). Among survivors, accounting for
the remaining 69.0% of the sample, brain stem (32.0%) and ventricle,
NOS (23.0%) were most prevalent locations. Mortality was greater
for brain, NOS (57.1%), temporal lobe (50.0%), and ventricle, NOS
(37.5%) compared to the overall average of 31.3%. There was one
case involving the cerebrum, which resulted in death. Lower rates
were observed for the brain stem, frontal lobe, and parietal lobe with
a single case for cerebellum surviving. A significant difference in
mortality was observed for brain, NOS versus other locations (57.1%
vs. 24.0%, p=0.094).
Pairwise comparisons among tumor locations (Table 3) revealed
a significantly lower mortality rate for brain stem compared to brain,
NOS (12.5% vs. 57.1%, p=0.067), and to cerebrum (12.5% vs. 100%,
p=0.047). Tumors located in the brain, NOS also resulted in greater
mortality than in frontal lobe (57.1% vs. 0.0%, p=0.091). Cerebral
tumors had higher rates of mortality if compared with both frontal
and parietal lobes (100% vs. 0.0% for both, p=0.046 and p=0.083),
respectively.
Figure 1
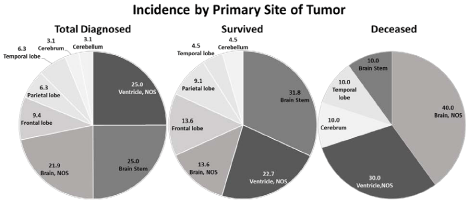
Figure 1
Comparative analysis of primary tumor sites in ependymoma total diagnosed patients vs. survived vs. deceased patients at the time of data analysis.
Total number of 32 cases were analyzed using R, ver. 2.15.1. Ependymoma was located in the ventricle NOS (25.0% vs. 22.7%, p=0.660), in brain stem (25.0%
vs. 31.8%, p=0.186), in brain, NOS (21.9% vs. 13.6%, p=0.094), in frontal lobe (9.4% vs. 13.6%, p=0.220), in parietal lobe (6.3% vs. 9.1%, p=0.325), in temporal
lobe (6.3% vs. 4.5%, p=0.555), in cerebrum (3.1% vs. 0.0%, p=0.132), and in cerebellum (3.1% vs. 4.5%, p=0.493), respectively.
Table 2
Table 2
Mortality rate overall and by location. Overall average for survivors was 68.8% of the sample. Among survivors, brain stem (32.0%) and ventricle, NOS (23.0%)
were most prevalent locations. Overall average for deceased patients was 31.3%. Among the deceased, mortality was greater for brain, NOS (57.1%), temporal lobe
(50.0%), and ventricle, NOS (37.5%). Cerebrum location (1 case) resulted in death. Lower rates of mortality were observed for the brain stem, frontal lobe, and parietal
lobe with a single case for cerebellum surviving. A significant difference in mortality was observed for brain, NOS versus other locations (57.1% vs. 24.0%, p=0.094).
Table 3
Table 3
Pairwise comparisons of mortality rates among locations. Test represents significantly lower mortality rate for brain stem compared to brain, NOS (12.5% vs.
57.1%, p=0.067), to cerebrum (12.5% vs. 100%, p=0.047), respectively. Greater mortality of brain, NOS was significant compared to locations in frontal lobe (57.1%
vs. 0.0%, p=0.091). Cerebrum showed significantly greater rates of mortality compared to frontal lobe (100% vs. 0.0%, p=0.046) and to temporal lobe (100% vs. 0.0%,
p=0.083), respectively.
Discussion
Physicians are currently trying to discern the most effective
treatments for ependymoma patients and identify associated
prognostic factors to better evaluate outcomes. Due to their rarity,
researchers must rely on a paucity of information that can only be
retrospectively analyzed.
Tumor site
Our data for a total diagnosed ependymoma population were
analyzed based on tumor primary site in living and deceased
patients who underwent treatments. It was found that treatment of
ependymoma located in the brain stem, frontal lobe, and parietal lobe
had significantly greater survival and recurrence-free outcomes than
tumors in other regions of the brain: cerebrum, cerebellum, temporal
lobe, brain NOS, or ventricles. These findings were supported by
McGuire et al. where cranial variants of ependymoma have a less
favorable outcome than primary spinal cord ependymomas [11]. It
has been reported that location within the spinal cord may also affect
outcome, with tumors in the lower portion of the spinal cord having
a worse prognosis [28].
Treatment modality and recurrence
Surgical excision with attempted GTR is the current gold standard
for ependymoma treatment. The administration of concurrent RT is
contingent upon the extent of resection, tumor grade, patient age,
and the presence or absence of tumor dissemination, which would
most commonly occur through the cerebrospinal fluid [12,29-34].
Since 80% of our patients who died underwent STR rather than GTR,
GTR could perhaps have been attempted on some of our diagnosed
patients. As was reported by other clinicians, STR is sometimes
preferred over GTR if the physician wishes to decrease the risk of
debilitating morbidity to the patient [8,28,35,36]. STR excises less
tissue and is therefore less likely to cause comorbidities. Based on our
observations, there seems to be a correlation between mortality and
surgery type in that GTR confers better prognosis.
Among the 8 patients who became disease-free with no
recurrence after treatment, 3 are now deceased. All three underwent
a combination of surgery with RT treatment and were diagnosed
with grade II tumors located in the brain stem (1 patient) and brain,
NOS (2 patients). Among the 5 living, 2 were diagnosed with grade II
tumors and underwent a combination of surgery and RT treatment.
In living individuals, the primary tumor sites were in the frontal lobes
(2 patients) and the brain, NOS (1 patient).
In this study, recurrence data were not reported for 11 patients,
but it is not unusual for patients whose symptoms have dissipated
to neglect contact with the healthcare system. Recurrence was seen/
reported in only 4 (12.5%) of our patients.
It has been reported that recurrence is relatively common in
grade III ependymomas and it is not uncommon with grade II
tumors [37]. Subependymomas are generally well-circumscribed
and well-differentiated, thus surgical resection with or without
concurrent radiotherapy is usually very effective in conferring a
favorable long-term prognosis [2,37,38]. It is known that survival
rates for subependymoma are generally higher than those of grade II
or grade III [10,39,40]. On the other hand, anaplastic ependymomas
most likely metastasize, recur, and diminish overall survival. Studies
published by several groups have shown anywhere from 42% to 100%
recurrence in grade III ependymoma [39,41-44]. In our observations,
3 patients with anaplastic ependymomas were still alive at the time of
analysis and had no recurrence. It is known that subependymomas
usually carry very different prognoses dependent on location and age
[18-21,45]. Therefore, we hypothesize that location and age might
contribute more to prognosis than histopathological qualities of the
tumor.
RT was administered in 50% of our ependymoma cases and was
excluded from treatment regimens of the pediatric group, but CT was
administered in only 12.5% of all cases. This approach corresponds
well with the current standard of ependymoma treatment and has
been reported in several other studies [26,45]. CT has not been
found to decrease instance of tumor recurrence and potentially even
exacerbates progression of the tumor by conferring natural selection,
and thereby resistant qualities upon the tumor cells [46,47]. Some
studies have shown that CT can delay progression and provide
palliative relief to patients with ependymoma, but it has not been
found to increase survival [25-27,41]. According to our findings, RT
also does not seem to significantly improve survival.
Age
Age associated observations in our study show that 65.6% of the
patients were adults and only 34.4% were children. Forty percent of
deceased patients were under 18 years of age and a survival curve
estimated a 65.6% of pediatric 5-year survival rate. These findings
corresponded with 5-year survival rates published by the American
Cancer Society (ACS) which demonstrate that ependymoma has the
4th worst rate among child brain tumors with a value of 75% (with
a pediatric qualifier of 19 years or younger) [48]. Our adult group
showed a 5-year survival rate of 78.4%. This may illustrate the recent
advances in cancer treatment.
Gender
The influence of gender has not been thoroughly investigated
in ependymoma studies because it almost equally afflicts men and
women and progresses similarly in both. However, in our study,
59.4% of all diagnosed patients were men and 40.6% were women.
Race
Another neglected survival relationship is ependymoma outcome
by race. Our study showed that despite the similarity in mortality
between white and other races, a correlation might exist between
ethnicity and ependymoma development since 71.9% of all patients
was white. However, all of the patients in our study were from Central
Texas. Thus, considering the local demographics (whites are less
predominant than Hispanics), the fact that more whites were afflicted
than any other race possibly denotes to genetic correlation, although
no genetic markers have been discovered as of yet.
Limitations and Conclusion
The purpose of this study is to add obtained knowledge to
currently available ependymoma literature. It is expected that the
field will benefit from additional information in this area to better
understand ependymoma associated prognostication. Although
the epidemiologic literature on brain tumors is inconclusive in
many areas, there is a pressing need for more researchers to study
ependymoma epidemiology.
The present study has several limitations: Even though this study
covers 37-year time frame which resulted in change of guidelines,
diagnostics, and treatment of disease, this study does not account for
the impact of time in respect to treatment of documented ependymoma
cases. This study has a limited sample size available from the Scott
& White Brain Tumor Registry. Despite the institutional reliability
and accuracy, all retrospective and exploratory investigations are
inherent to limitations including variability of diagnostic criteria and
tools, lack of diagnosis confirmation, and loss to follow up. We did
not include tumor grade/stage in our analysis which may possibly be
a confounder or effect modifier. However, since CNS tumor grading
and staging is continuously subject to change over time, age, race
and location of the primary tumor are likely to be more important
prognostic indicators in current consideration. Finally, the ethnical
diversity of the Central Texas population should be taken into
account as it may not be representative of the overall US population.
In conclusion, our study suggests that one of the major factors
that can be used to evaluate prognosis of ependymoma patients is
primary site of the tumor. Tumor locations in the brain stem, frontal
lobe, and parietal lobe seem to have greater survival outcomes and
lower recurrence of disease when treated surgically at the very least,
when compared to other regions of the brain. There also seems to
be a genetic correlation of ependymoma development with white
ethnicity, and male sex.
Acknowledgement
This work was supported, in part, by NIH-R01-NS-067435 (JHH) and Baylor Scott & White Healthcare Plummer Chair’s Fund (JHH).
References
- Vaidya K, Smee R, Williams JR. Prognostic factors and treatment options for pediatric ependymomas. J Clin Neurosci. 2012; 19: 1228-1235.
- Massimino M, Giangaspero F, Garrè ML, Genitori L, Perilongo G, Collini P, et al. Salvage treatment for childhood ependymoma after surgery only: pitfalls of omitting “at once” adjuvant treatment. Int J Radiat Oncol Biol Phys. 2006; 65: 1440-1445.
- Duffner, Krischer JP, Sanford RA, Horowitz ME, Burger PC, Cohen ME, et al. Prognostic factors in infants and very young children with intracranial ependymomas. Pediatr Neurosurg. 1998; 28: 215-222.
- Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001; 17: 503-511.
- Rodríguez D, Cheung MC, Housri N, Quinones-Hinojosa A, Camphausen K, Koniaris LG. Outcomes of malignant CNS ependymomas: an examination of 2408 cases through the Surveillance, Epidemiology, and End Results (SEER) database (1973–2005). J Surg Res. 2009; 156: 340-351.
- Villano JL, Parker CK, Dolecek TA. Descriptive epidemiology of ependymal tumours in the United States. Br J Cancer. 2013; 108: 2367-2371.
- Primary brain tumors in the United States: statistical report, 1995-1999. CBTRUS; 2002.
- Zacharoulis S, Moreno L. Ependymoma: an update. J Child Neurol. 2009; 24: 1431-1438.
- Purdy E, Johnston DL, Bartels U, Fryer C, Carret AS, Crooks B, et al. Ependymoma in children under the age of 3 years: a report from the Canadian Pediatric Brain Tumour Consortium. J Neurooncol. 2014; 117: 359-364.
- Amirian ES, Armstrong TS, Aldape KD, Gilbert MR, Scheurer ME. Predictors of survival among pediatric and adult ependymoma cases: a study using Surveillance, Epidemiology, and End Results Data from 1973 to 2007. Neuroepidemiology. 2012; 39: 116-124.
- McGuire CS, Sainani KL, Fisher PG. Incidence patterns for ependymoma: a surveillance, epidemiology, and end results study. J Neurosurg. 2009; 110: 725-729.
- Aizer AA, Ancukiewicz M, Nguyen PL, Macdonald SM, Yock TI, Tarbell NJ, et al. Natural history and role of radiation in patients with supratentorial and infratentorial WHO grade II ependymomas: results from a population-based study. J Neurooncol. 2013; 115: 411-419.
- Horn B, Heideman R, Geyer R, Pollack I, Packer R, Goldwein J, et al. A multi-institutional retrospective study of intracranial ependymoma in children: identification of risk factors. J Pediatr Hematol Oncol. 1999; 21: 203-211.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvett A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (Berl). 2007; 114: 97-109.
- Merchant TE, Haida T, Wang MH, Finlay JL, Leibel SA. Anaplastic ependymoma: treatment of pediatric patients with or without craniospinal radiation therapy. J Neurosurg. 1997; 86: 943-949.
- Phi JH, Wang KC, Park SH, Kim IH, Kim IO, Park KD, et al. Pediatric infratentorial ependymoma: prognostic significance of anaplastic histology. J Neurooncol. 2012; 106: 619-626.
- McLaughlin MP, Marcus RB Jr, Buatti JM, McCollough WM, Mickle JP, Kedar A, et al. Ependymoma: results, prognostic factors and treatment recommendations. Int J Radiat Oncol Biol Phys. 1998; 40: 845-850.
- Rushing EJ, Cooper PB, Quezado M, Begnami M, Crespo A, Smirniotopoulos JG, et al. Subependymoma revisited: clinicopathological evaluation of 83 cases. J Neurooncol. 2007; 85: 297-305.
- Hou Z, Wu Z, Zhang J, Zhang L, Tian R, Liu B, et al. Clinical features and management of intracranial subependymomas in children. J Clin Neurosci. 2013; 20: 84-88.
- Ragel BT, Osborn AG, Whang K, Townsend JJ, Jensen RL, Couldwell WT. Subependymomas: an analysis of clinical and imaging features. Neurosurgery. 2006; 58: 881-890.
- Scheithauer BW. Symptomatic subependymoma: report of 21 cases with review of the literature. J Neurosurg. 1978; 49: 689-696.
- Perilongo G, Massimino M, Sotti G, Belfontali T, Masiero L, Rigobello L, et al. Analyses of prognostic factors in a retrospective review of 92 children with ependymoma: Italian Pediatric Neuro-Oncology Group. Med Pediatr Oncol. 1997; 29: 79-85.
- Pollack IF, Gerszten PC, Martinez AJ, Lo KH, Shultz B, Albright AL, et al. Intracranial ependymomas of childhood: long-term outcome and prognostic factors. Neurosurgery. 1995; 37: 655-667.
- Kim SK, Lim SY, Wang KC, Kim YY, Chi JG, Choi YL, et al. Overexpression of cyclooxygenase-2 in childhood ependymomas: role of COX-2 inhibitor in growth and multi-drug resistance in vitro. Oncol Rep. 2004; 12: 403-409.
- Timmermann B, Kortmann RD, Kühl J, Meisner C, Slavc I, Pietsch T, et al. Combined postoperative irradiation and chemotherapy for anaplastic ependymomas in childhood: results of the German prospective trials HIT 88/89 and HIT 91. Int J Radiat Oncol Biol Phys. 2000; 46: 287-295.
- Robertson PL, Zeltzer PM, Boyett JM, Rorke LB, Allen JC, Geyer JR, et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children’s Cancer Group. J Neurosurg. 1998; 88: 695-703.
- Evans AE, Anderson JR, Lefkowitz-Boudreaux IB, Finlay JL. Adjuvant chemotherapy of childhood posterior fossa ependymoma: cranio-spinal irradiation with or without adjuvant CCNU, vincristine, and prednisone: a Childrens Cancer Group study. Med Pediatr Oncol. 1996; 27: 8-14.
- Safaee M, Oh MC, Mummaneni PV, Weinstein PR, Ames CP, Chou D, et al. Surgical outcomes in spinal cord ependymomas and the importance of extent of resection in children and young adults: Clinical article. J Neurosurg Pediatr. 2014; 13: 393-399.
- Korshunov A, Golanov A, Sycheva R, Timirgaz V. The histologic grade is a main prognostic factor for patients with intracranial ependymomas treated in the microneurosurgical era: an analysis of 258 patients. Cancer. 2004; 100: 1230-1237.
- Rogers L, Pueschel J, Spetzler R, Shapiro W, Coons S, Thomas T, et al. Is gross-total resection sufficient treatment for posterior fossa ependymomas? J Neurosurg. 2005; 102: 629-636.
- Merchant TE, Li C, Xiong X, Kun LE, Boop FA, Sanford RA. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol. 2009; 10: 258-266.
- Mansur DB, Perry A, Rajaram V, Michalski JM, Park TS, Leonard JR, et al. Postoperative radiation therapy for grade II and III intracranial ependymoma. Int J Radiat Oncol Biol Phys. 2005; 61: 387-391.
- English MW. 21st century management of ependymoma. Lancet Oncol. 2009; 10: 206-207.
- Awaad YM, Allen JC, Miller DC, Schneider SJ, Wisoff J, Epstein FJ. Deferring adjuvant therapy for totally resected intracranial ependymoma. Pediatr Neurol. 1996; 14: 216-219.
- Ideguchi M, Kajiwara K, Yoshikawa K, Sadahiro H, Nomura S, Fujii M, et al. Characteristics of intraoperative abnormal hemodynamics during resection of an intra-fourth ventricular tumor located on the dorsal medulla oblongata. Neurol Med Chir (Tokyo). 2013; 53: 655-662.
- Li TY, Chu JS, Xu YL, Yang J, Wang J, Huang YH, et al. Surgical strategies and outcomes of spinal ependymomas of different lengths: analysis of 210 patients: clinical article. J Neurosurg Spine. 2014; 21: 249-259.
- Preston-Martin S. Epidemiology of primary CNS neoplasms. Neurol Clin. 1996; 14: 273-290.
- Armstrong TS, Vera-Bolanos E, Gilbert MR. Clinical course of adult patients with ependymoma: results of the Adult Ependymoma Outcomes Project. Cancer. 2011; 117: 5133-5141.
- Merchant TE, Jenkins JJ, Burger PC, Sanford RA, Sherwood SH, Jones-Wallace D, et al. Influence of tumor grade on time to progression after irradiation for localized ependymoma in children. Int J Radiat Oncol Biol Phys. 2002; 53: 52-57.
- Roldán Urgoiti GB, Singh AD, Tsang RY, Nordal RA, Lim G, Chan JA, et al. Population based analysis of ependymoma patients in Alberta from 1975 to 2007. Can J Neurol Sci. 2014; 41: 742-747.
- Massimino M, Gandola L, Giangaspero F, Sandri A, Valagussa P, Perilongo G, et al. Hyperfractionated radiotherapy and chemotherapy for childhood ependymoma: final results of the first prospective AIEOP (Associazione Italiana di Ematologia-Oncologia Pediatrica) study. Int J Radiat Oncol Biol Phys. 2004; 58: 1336-1345.
- Oya N, Shibamoto Y, Nagata Y, Negoro Y, Hiraoka M. Postoperative radiotherapy for intracranial ependymoma: analysis of prognostic factors and patterns of failure. J Neurooncol. 2002; 56: 87-94.
- Tarapore PE, Modera P, Naujokas A, Oh MC, Amin B, Tihan T, et al. Pathology of spinal ependymomas: an institutional experience over 25 years in 134 patients. Neurosurgery. 2013; 73: 247-255.
- Tihan T, Zhou T, Holmes E, Burger PC, Ozuysal S, Rushing EJ. The prognostic value of histological grading of posterior fossa ependymomas in children: a Children’s Oncology Group study and a review of prognostic factors. Mod Pathol. 2008; 21: 165-177.
- Bloom HJ, Glees J, Bell J, Ashley SE, Gorman C. The treatment and long-term prognosis of children with intracranial tumors: a study of 610 cases, 1950-1981. Int J Radiat Oncol Biol Phys. 1990; 18: 723-745.
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008; 8: 592-603.
- Fischer C, Haque SS, Huse JT, Blochin E, Souweidane MM, Lis E, et al. Extraneural ependymoma: Distant bone, lung, liver, and lymph node metastases following bevacizumab. Pediatr Blood Cancer. 2013; 60: 143-5.
- Cancer in children & adolescents. Atlanta: American Cancer Society. 2014.