Review Article
Gangliocytomas and Gangliogliomas: Review of Clinical, Pathologic and Genetic Features
Yazmin Odia*
Neuro-Oncology, Miami Cancer Institute, Baptist Health South Florida, USA
*Corresponding author: Yazmín Odia, MD MS, Neuro-Oncology Miami Cancer Institute, Baptist Health South Florida, 8940 N Kendall Drive, Suite 300E Miami FL 33176, USA
Published: 14 Jun, 2016
Cite this article as: Odia Y. Gangliocytomas and
Gangliogliomas: Review of Clinical,
Pathologic and Genetic Features. Clin
Oncol. 2016; 1: 1017.
Abstract
Gangliocytomas and Gangliogliomas are well-differentiated, slowly growing neuroepithelial
tumors, composed of neoplastic, mature ganglion cells alone or in combination with neoplastic glial
cells, respectively. Surgical resection is the standard of care, radiation used for malignant features
or unresectable tumors. The epidemiology, clinical, pathologic and genetic features of these rare
primary brain tumors are described.
Keywords: Gangliocytomas; Gangliogliomas; Review; Chromosomal imbalances; CDKN2A
Introduction
Historical perspective
Gangliocytomas and gangliogliomas are well-differentiated, slowly growing neuroepithelial
tumors, composed of neoplastic, mature ganglion cells alone or in combination with neoplastic glial
cells, respectively [1]. "Gangliogliomas" were first described by James Ewing in 1926 and adopted
by Cyril B. Courville in 1930 [2]. According to the 2007 World Health Organization (WHO)
classification, both gangliogliomas and gangliocytomas are benign tumors (WHO grade I), [1] but
malignant transformations to anaplastic ganglioglioma, WHO grade III, have been reported [3]. In
large series, WHO grade I, II and III tumors comprise 86%, 9% and 5% of all gangliogliomas, while
<1% show features consistent with glioblastoma (WHO grade IV) [4,5].
The benign (WHO grade I) dysplastic gangliocytoma of the cerebellum (also called LhermitteDulcos)
was first described by J. Lhermitte and P. Duclos and Spiegel in 1920, [6] but only recently
recognized as part of Cowden syndrome. Cowden syndrome or multiple hamartoma-neoplasia
syndrome, is an uncommon autosomal dominant disorder characterized by mucocutaneous lesions
and systemic malignanciesc [1,7].
Epidemiology
Ganglioglioma and gangliocytomas together comprise 0.4% of all CNS and 1.3% of all brain
tumors based on larger surgical series [1,8,9]. They comprise the most frequent type of glioneuronal
tumors (38.5%) [10]. Second only to benign cortical malformations, gangliogliomas comprise 37-
46% of all tumors associated with intractable epilepsy in surgical series. The age at presentation
ranges from 2 months to 70 years, with a mean/median age from 8.5 to 25 years. There is a slight
male to female predominance (1.1-1.9 to 1) [1,11-14]. Dysplastic gangliocytomas of the cerebellum
are too rare to permit demographic characterization [1].
Location and presentation
Gangliocytomas and gangliogliomas predominantly arise in the cerebral hemispheres (up to
70%), but also occur in the cerebellum (15-17%), ventricles (~3%), brainstem (~3%), spinal cord
(as low as 2-3%) and extra-axial sites (~1%), such as the optic chiasm or cerebellopontine angle
[1,10]. Cerebral tumors predominantly localize to the temporal (40-75%), followed by frontal
(12-15), parietal (6-7%) or occipital (~3%) lobes and infrequently multilobar (6-7%). Meningeal
or leptomeningeal spread is rarely reported [13,15]. The distinct entity dysplastic gangliocytoma
of the cerebellum (Lhermitte-Duclos) strictly arises from the cerebellum, usually confined to a
hemisphere, but occasionally multifocal [1].
Clinical symptoms vary depending on location. Given their predilection for temporal and
frontal lobes, new onset seizures and refractory epilepsy are their most common presentation.
Aside from seizures (50-90%), other common symptoms include increased intracranial pressure
(40-50%), cerebellar signs (33%) and focal neurologic deficits (12-15%). Rare symptoms include
memory disturbances, cranial nerve palsies and psychiatric symptoms (1-7%) [10]. Dysplastic
gangliocytomas of the cerebellum, as expected, present with cerebellar
signs and symptoms of mass effect, including hydrocephalus [1,11].
Imaging findings
On computed tomography (CT), gangliocytomas and
gangliogliomas appear as well-circumscribed masses in 80%, with
predominant solid (45-50%) or solid cystic (25-30%) features and
often a mural nodule (25%) (Figure 1). Focal calcifications are
frequent (40-50%), while mass effect and vasogenic edema are found
in <10% of cases [10]. Contrast enhancement is also common,
though often faint and rarely absent. Scalloping of the adjacent
calvarium may occur. Magnetic resonance imaging (MRI) reveals T1
hypointense and T2 hyperintense circumscribed mass lesions, with
variable enhancement from none to marked and solid, rim or nodular
[16,17]. Dysplastic gangliocytomas of the cerebellum share imaging
characteristics, namely solid-cystic features, no or faint enhancement
and calcifications. A gyriform 'tiger-striped' appearance in a unilateral
cerebellar hemisphere is the hallmark finding on MRI [7]. The relative
low cellularity of gangliogliomas correlates with high apparent
diffusion coefficient (ADC) values compared to low and high-grade
gliomas [18]. Figure 2 depicts typical MRI and head CT findings.
Figure 1
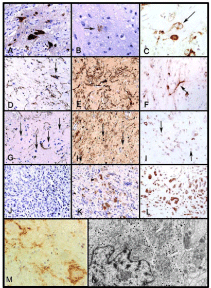
Figure 1
Representative Pathology of Gangliogliomas and Gangliocytomas.
The benign component of a lesion contains large NeuN-positive neurons
(A, 400×), which occasionally stained for chromogranin (arrow) within the
neuronal soma (B, 630×), with some being binucleate and nestin positive
(C, arrow, 400×). The large GFAP-positive astrocytes (D, 400×) were also
S-100 positive (E, 400×) and nestin positive (F, 400×). Between the large
glial and neuronal cells were additional stromal cells with small rounded
nuclei (arrows) that did not stain for GFAP (G, 400×) but did stain positive
for both S-100 (H) and nestin (I). The malignant component of the tumor
was composed of small cells in a loose myxoid stroma and did not stain
for GFAP (J, 400×) but demonstrated variable nuclear and cytoplasmic
reactivity for S-100 (K, 400×) and strong reactivity for nestin (L, 400×).
Immunostaining with antibody to synaptophysin is seen (M). The astrocytes
show no staining; some coarse granular immunoreactivity in the neuropil
background is present, but there is striking staining along the surfaces of
the large neoplastic neurons. This pattern is characteristic of gangliogliomas.
Ultrastructure of ganglioglioma: A neuronal cell containing a large number
of dense core granules, well developed rough endoplasmic reticulum, and
Golgi complexes is shown. Two axonal boutons form synapses with the cell
membrane (N, 12, 600x). Figures reprinted and modified from A Pandita et al,
Neuro-Oncology 2007:9(2) (A-L), DC Miller et al, 1993:79 (M) and T Hirose
et al, 1996:79(5) (N) with permission from Oxford University Press, JNS
Publishing Group, and John Wiley and Sons, respectively.
Histopathology
Macroscopically, gangliogliomas are solid or cystic masses.
Calcification is common in gross specimens, while hemorrhage and
necrosis are rare. Dysplastic gangliocytomas of the cerebellum display
discrete regions of hypertrophy and course gyral patterns deep in the
cerebellar hemisphere [1].
Microscopically, gangliocytomas are characterized by irregular
arrangement of neoplastic ganglion cells, defined as large, multipolar
neurons with dysplastic features. The stroma consists of nonneoplastic
glial elements, commonly with a dense perivascular
reticulin network. Most are at least focally fibrous with collagen
staining in the background. Gangliogliomas share the histopathologic
findings of gangliocytomas, plus an admixture to varying degrees of
neoplastic glial elements. The glial component is typically astrocytic,
either fibrillary or pilocytic. Oligodendroglial "fried-egg" or
ependymal "perivascular pseudorosettes" patterns are exceedingly
rare. Rosenthal fibers and granular bodies are occasionally associated
with the astrocytic component. Gangliogliomas are best differentiated
from low grade gliomas by four hallmark findings. Clusters of large
cells potentially representing neurons are required for diagnosis.
Neoplastic glial cells should not cluster around the neoplastic
neurons. Fibrosis (desmoplasia) and calcification are the remaining
two defining features. Binucleate neurons are diagnostic, but evident
in <50% of cases. Lymphocytic or plasma cell infiltrates are common
but nonspecific features [4,19].
Anaplastic gangliogliomas typically consist of malignant
transformation of the glial component and defined by increased
mitotic activity, prominent micro-vascular proliferation, necrosis,
and high MIB-1 and TP53 labeling indices, consistent with their
WHO grade III classification. Most gangliogliomas have low
immunoreactivity for MIB-1 marker of cellular proliferation (mean
1.1 +/- 1.0) and Ki-67 (<1%) labeling indices, consistent with their
WHO grade I classification. Anaplastic gangliogliomas, in contrast,
have significantly higher MIB-1 (as high as 10.2) and Ki-67 (up to
10%) labeling indices, which have also been correlated with increased
size of ganglion cells [11,13,14]. Mitoses are rare in cells with neuronal
appearance, [8,11,13,19] while proliferation indices, as denoted
by the Ki-67 nuclear antigen, is exclusively found in the astrocytic
component. Malignant transformation of both neuronal and glial
components remains exceedingly rare [20-22].
The ganglion cells in dysplastic gangliocytomas of the
cerebellum causes diffuse enlargement of the molecular and internal
granular layers of the cerebellum. The cerebellar architecture is
characteristically preserved, with distorted and enlarged but intact
folia. Parallel arrays of abnormal myelinated axon bundles are
common in the outer molecular layer. Granule neurons can be found
under the pia or molecular layer, while Purkinje cells are sparse or
absent. Calcification and ecstatic vessels are common, while vacuoles
in the molecular layer and white matter are rare [1].
Immunohistochemistry is essential to the accurate diagnosis of
gangliocytomas and gangliogliomas. In particular synaptophysin
and glial fibrillary acidic protein (GFAP) stains define the relative
neuronal and glial components, respectively [19]. Synaptophysin
staining along the surface of large neoplastic neurons is characteristic
of gangliogliomas, but coarse granular immunoreactivity in the
neuropil background is also common [19]. Neoplastic neurons
also show immunoreactivity for Class 3 betatubulin (100%),
neurofilament protein (NFP, 90%), chromogranin A (86%) and
S-100 protein (71%), less frequently for vimentin (24%) [11]. Glial
cells, in turn, are immunoreactive for GFAP, S-100 protein and
vimentin neuroepithelial markers [11]. Microtubule-associated
protein-2 (MAP2) immunoreactivity, a hallmark of most low grade
glial tumors, is virtually absent in the neoplastic glial component of
Gangliogliomas [23].
Ultrastructurally, gangliogliomas show large neuronal cells with
cell bodies contacted by numerous neuritic endings containing small
clear synaptic vesicles, corresponding to the diagnostic synaptophysin
staining pattern. Neuronal cells characteristically contain numerous,
dense core granules within the cell body and processes [11]. Spherical
protein bodies characteristic for catecholamine neurons in humans
have also been reported in select ganglioglioma cases [24]. Distal
from the cell bodies, electron microscopy (EM) reveals a dense
neuropil of neuritis with microtubules, intermediate filaments and
other organelles, but no synaptic endings or dendritic spines. The
admixed glial processes are filled with denser filament without parallel
microtubules. The glial nuclei are darker with more homogeneous
chromatin than neoplastic neuronal nuclei [1,19].
Figure 2
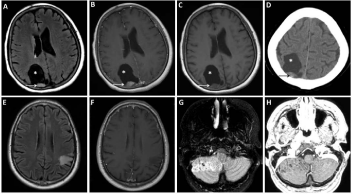
Figure 2
Representative Imaging Findings of Gangliogliomas and Gangliocytomas.
The solid and cystic (*) and enhancing (B-C) and partly calcified (D) mural nodule (arrow) are common findings of gangliogliomas as depicted in T2/FLAIR (A)
and post- (B) and pre-gadolinium T1 (C) MRI images and noncontrast head CT (D). Gangliocytomas are also often solid with semi-defined margins with no
corresponding enhancement as depicted in T2/FLAIR (E) and post-gadolinium T1 (F) MRI images. Dysplastic gangliocytomas of the cerebellum (Lhermitte-
Duclos) have a pathopneumonic MRI appearance of gyriform ‘tiger-striped’ appearance in a unilateral cerebellar hemisphere (G-H, T2/FLAIR and post-gadolinium
T1 MRI, respectively).
Pathogenesis and Molecular Genetics
One-third of gangliogliomas have chromosomal imbalances,
particularly gain of 7 or partial loss of 9q [15,25-27]. Molecular genetic
analysis of a ganglioglioma in a toddler with diffused leptomeningeal
involvement also revealed loss of chromosome 17p. An abnormal
karyotype strongly predicts adverse outcomes [3,15,20].
Mutations in key cell cycle regulators have also been associated
with gangliogliomas. Homozygous deletion of 9p21 including the
cyclin-dependent kinase inhibitor 2A (CDKN2A, p16Ink4A) gene, a
tumor suppressor involved in regulating the cell cycle, was detected in
10% of all gangliogliomas and two-thirds of anaplastic Gangliogliomas
[28,29]. There is growing evidence suggesting a functional link
between CDKN2A and BRAF genes. Concurrent CDKN2A loss and
BRAF activation has been associated with gliomagenesis in mouse
models, while the BRAF-V600E activating mutation, historically
associated with melanoma, colon and papillary thyroid carcinoma,
can induce CDKN2A. The BRAF-V600E mutation activates the RAS/
RAF/MEK/ERK signaling cascade implicated in various malignancies.
Not surprisingly, recent studies detected the BRAF-V600E mutation
in 20-50% of gangliogliomas 20-50%, more frequently in anaplastic
(50%) than in WHO grade I (18%) Gangliogliomas [28,29].
Phosphatidylinositol 3-kinases (PI3K) are also vital in cell growth,
proliferation, differentiation, motility, survival and intracellular
trafficking. While mutations in the PI3K pathway are associated with
glioma pathogenesis, no mutations were detected in downstream
effectors of the P13K pathway, specifically Ezrin, radixin and moesin
(ERM) genes in a large series of gangliogliomas. Higher expression of
most phosphorylated components of the PI3K/Akt/mTOR pathway,
including ERMs, PDK1, mTOR, E-BP1, EIF4G, ribosomal protein
S6 kinase phosphorylated at threonine 389 and 229 and ribosomal
protein S6, was detected in gangliogliomas when compared to normal
cortex [30,31].
While common in gliomas, IDH1-R132 and PTEN mutations
as well as CDK4 and EGFR amplification are notably absent in
gangliogliomas [31]. The IDH1-R132 mutation, the hallmark of
secondary glioblastomas, was reported in oligodendrogliomas with
ganglioglioma-like features (GGLF). With a clinical course more like
infiltrative gliomas than gangliogliomas, the GGLF actually reflects
neoplastic glial cells with extensive ganglioid differentiation given
the frequent 1p19q molecular signature on FISH and IDH1-R132
immunoreactivity in both glial and ganglioid cells [32]. TP53
mutations, detected in the majority of gliomas, are only rarely
reported in gangliogliomas with local recurrence or transformation
to glioblastoma (WHO grade IV) [31,33]. Mutational screening
of the NBN gene, commonly co-expressed with TP53 mutations in
medulloblastoma, found a single ganglioglioma with a c.511A>g
(p.Ile171Val) substitution on one allele of the NBN gene. Mutations
in the NBN (previously named NBS1) gene define the Nijmegen
breakage syndrome (NBS), a rare autosomal recessive chromosomal
instability disorder characterized by microcephaly, dysmorphic
features, immunodeficiency, radiosensitivity and increased risk
of cancers. Heterozygous NBN mutations are also associated with
increased risk of various neoplasms, including melanoma, nonHodgkin's
lymphomas, acute lymphoblastic leukemia, stomach and
colorectal cancer, breast and ovarian cancers, rhabdomyosarcoma
and medulloblastoma [34].
Cortical architectural abnormalities, including cortical dysplasia
and microdysgenesis, are found in 50% of gangliogliomas, near
but clearly separate from the tumor. The high incidence of cortical
malformations suggests aberrant development as a basis of their
pathogenesis [13]. Aberrant overexpression of ERMs effectors
of the PI3K pathway is evident in both cortical dysplasia and
Gangliogliomas [35]. Analysis of genes in the Reelin signaling cascade
involved in neuronal development failed to detect mutations in the
cyclin-dependent kinase CDK5, doubcortin DCX, p35 and disabled-1,
DAB-1 [36,37]. The TSC1 and TSC2 genes also function in cortical
differentiation and growth control. Tuberous sclerosis 2 (TSC2)
alterations, including polymorphisms in intron 4 and exon 41, are
overrepresented in patients with gangliogliomas, again suggesting
alterations in neuro-developmental signaling cascades [36,38]. TSC1
and TSC2 as well as PTEN genes have been molecularly linked to the
serine-threonine kinase (LKB1) gene, a master kinase involved in the
control of cell cycle arrest, p53-mediated apoptosis, WNT and TGFbeta
signaling, Ras-induced transformation, energy metabolism and
cell polarity. Peutz-Jeghers syndrome (PJS), an autosomal dominant
disorder, characterized by the benign hamartomatous polyps in the
gut and hyper-pigmented macules on the lips and oral mucosa, results
from inactivating germline mutations in the LKB1. While a few cases
of PJS with a germline LKB1 mutation developed a ganglioglioma, no
LKB1 gene mutations have been reported in sporadic Gangliogliomas
[39,40].
Various rare genetic syndromes have been associated with
aggressive gangliogliomas. A young child with stigmata of
neurofibromatosis type 2 (NF2) developed a rapidly growing,
exophytic intramedullary ganglioglioma at the cervicomedullary
junction. NF2 patients often develop aggressive malignant gliomas,
suggesting a causal link between the predilection for the cooccurrence
in this single case of the exceedingly rare NF2 syndrome
and a ganglioglioma [41]. A woman with Turner Syndrome (TS,
monosomy X) developed an anaplastic supratentorial ganglioglioma
(WHO grade III). After partial resection and involved field radiation
to 60 Gy, a gross total resection of local tumor recurrence revealed
transformation to a glioblastoma (WHO grade IV) that resulted
in her death 23 months after her initial diagnosis[42]. A child
with familial mild learning disability, characterized by congenital
cataract and developmental and speech delay, developed a metastatic
ganglioglioma with anaplastic transformation by the age of 2 years.
Mutational analysis revealed a unique germline 9q34.4 constitutional
tandem duplication resulting in breakpoints in intron 1 of TRAF2
and intron 16 of the EHMT1 gene. The result is a fusion transcript
encoding a truncated form of EHMT1. The ganglioglioma showed
complex chromosomal aberrations with further duplication of the
dub9q34 [43]. Other microdeletions, duplications and translocations
of the 9q32-qter region are associated with various pediatric cases
with neurodevelopmental disorders as described by Kleefstra et
al. [44]. These cases highlight the significant association between
chromosomal instability and gangliogliomas [45].
Cowden syndrome is an autosomal dominant disorder defined
by mucocutaneous lesions, specifically multiple trichilemmoma
(skin differentiation into the outer root sheath of the hair follicle)
and fibromas, as well as systemic malignancies, namely thyroid
neoplasms, breast carcinoma and hamartomatous polyps of the
colon [1,7]. Cowden syndrome is characterized by germline
PTEN mutations on chromosome 10q23 in virtually all adultonset
dysplastic gangliocytomas of the cerebellum (LhermitteDuclos).
Childhood-onset Lhermitte-Duclos lack PTEN mutations,
suggesting a distinct etiology. In 75% of Lhermitte-Duclos samples,
immunohistochemistry reveals complete or partial loss of PTEN
expression accompanied by elevated phosphorylated Akt, specifically
in the dysplastic gangliocytoma cells. Loss of PTEN function seems
sufficient to cause Lhermitte-Duclos as part of Cowden syndrome
even without the systemic findings typical of Cowden syndrome [46].
Microarray RNA expression in epilepsy-associated
gangliogliomas was compared to postmortem temporal lobe samples
from patients with seizure or other neurologic disorders. Microarray
results were validated by RT-PCR analysis of 11 selected genes and
immunostaining of involved proteins. This microarray analysis yielded
numerous target genes, but does not permit precise conclusions of
the specific role of these gene expression changes in the pathogenesis
or epileptogenicity of gangliogliomas. Specifically, gangliogliomas
highly expressed genes involved various in immune and inflammatory
responses, including class II histocompatibility antigens, interleukins
and their receptors, as well as genes involved in the TGF-beta,
Toll-like receptor and T-cell receptor signaling pathways. Both the
classical and alternative complement pathways as well as components
of the coagulation cascade and iron ion homeostasis were also
overexpressed. Synaptic transmission was the most prominent
underexpressed process, including voltage gated potassium and
calcium channels as well as sodium channel subunits. GABA signaling
pathway was downregulated via suppresion of GABA-A receptors
and associated proteins as well as increased expression of the sodiumpotassium
chloride co-transporter (NKCC1), known to modulate
the GABA receptor-mediated response. Alterations in these ion and
GABA channels may underlie the epileptogenicity of gangliogliomas.
Several gene expression changes were specific to gangliogliomas in
contrast to other epileptogenic tissues. Genes involved in extracellular
matrix and cell adhesion were significantly activated. Additional
genes associated with development, cell cycle and Wnt-1/betacatenin
signaling pathway. Genes regulating angiogenesis, including
fibroblast growth factors as well as angiopoietin, angiogenin and
neuropilin, are prominently overexpressed [47].
Treatment and Clinical Outcomes
Gangliogliomas are typically benign tumors, while less than 5%
show WHO grade III pathology. Good prognostic factors include
temporal localization, complete surgical resection and long-standing
epilepsy [48]. Survival inversely correlates with WHO grade [49].
Based on the SEER registry, median overall survival for anaplastic
gangliogliomas is 28.5 months, where only debulking surgery (93%)
and extent of disease predict outcome [50]. The 5-year survival rates
also differ significantly based on localization, 93%, 84% and 33% for
cerebral, spinal cord and brainstem gangliogliomas, respectively. The
progression free rates at 5-year are 95% and 36% for cerebral and
spinal cord tumors, while only 53% of brainstem gangliogliomas are
progression free at 3 years [12,51]. In large surgical series, seizure
control is reached in at least 85% of gangliogliomas, up to 96% after
gross total but only 54% after subtotal resectionc [52]. Thus, gross total
resection, whenever safe, remains the mainstay and the only curative
treatment for gangliogliomas, whether low-grade or malignant [53].
Given the low recurrence rate of low grade gangliogliomas and
the unclear benefit of additional treatment after surgical resection,
radiation is generally reserved for incompletely resected tumors,
especially those with any evidence of malignant features. Yet, based
on the SEER registry, only 36% of anaplastic gangliogliomas received
adjuvant radiotherapy. The SEER registry analysis revealed no impact
of radiotherapy on overall survival [50]. A meta-analysis of 402
gangliogliomas revealed radiation therapy improved local control
but failed to impact overall survival in both low and high grade
gangliogliomas. Furthermore, the benefit of radiation therapy was
only evident after subtotal not gross total resections [53].
Anaplastic change of the glial component predicts a shorter time
to recurrence, greater risk of recurrence or malignant transformation,
but variably impacts overall survival [8,9,11-13]. Transformation
of both glial and neuronal component is exceedingly rare [22].
Transformation to anaplastic (WHO grade III) from low grade
gangliogliomas has been reported after incomplete resection and/or
decades after involved field radiation. Transformation to glioblastoma
(WHO grade IV) is rare and exclusive to partially resected anaplastic
gangliogliomas years-to-decades after radiotherapy [42,54]. Only a
few cases of anaplastic gangliogliomas unrelated to radiotherapy have
been reported [5].
Summary
Gangliocytomas and gangliogliomas are well-circumscribed
indolent neuroepithelial tumors. Their prognosis is generally good,
even curable with complete resection in the majority of cases. While
usually unresectable, dysplastic gangliocytoma of the cerebellum
(Lhermitte-Duclos) typically has a benign course. However, they
commonly coexist with less favorable multiple hamartoma-neoplasms
as part of Cowden syndrome. Primary anaplastic ganglioglioma are
rare, while malignant transformation to anaplastic ganglioglioma
or glioblastoma often occurs years-to-decades after radiation.
Malignant features are almost exclusive to the neoplastic glial rather
than ganglion cells. Aggressive features include leptomeningeal
dissemination on imaging as well as anaplasia and brisk mitoses on
histopathology.
Distinct genetic abnormalities suggest multiple etiologies for
this subgroup of glioneuronal tumors. Chromosomal imbalances are
evident in a third of gangliogliomas, while CDKN2A gene deletion
is detected in two-thirds of anaplastic gangliogliomas. Cowden
syndrome, an autosomal dominant disorder characterized by
germline PTEN mutations, defines all adult-onset cases of LhermitteDuclos.
Other rare genetic syndromes associated with gangliogliomas,
particularly with aggressive clinical or pathology features, include
NF2, Peutz-Jager and Turner syndromes. Alterations in genes
involved in neurodevelopment pathways and the co-existence of
cortical architectural abnormalities remote from the primary tumor
implicates aberrant cortical development in the pathogenesis of
gangliogliomas.
References
- Louis D, Ohgaki H, Wiestler O, Cavenee W, Burger PC, Jouvet A, et al. WHO Classification of tumours of the central nervous system. Lyon: IARC; 2007.
- Courville CB. Gangliocytoma myelinicum diffusum of the cerebellar cortex; review of the literature and report of case. Bull Los Angel Neuro Soc. 1958; 23: 72-80.
- Jay V, Squire J, Blaser S, Hoffman HJ, Hwang P. Intracranial and spinal metastases from a ganglioglioma with unusual cytogenetic abnormalities in a patient with complex partial seizures. Childs Nerv Syst. 1997; 13: 550-555.
- Blumcke I, Wiestler O. Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol. 2002; 61: 575- 584.
- DeMarchi R, Abu-Abed S, Munoz D, Loch Macdonald R. Malignant ganglioglioma: case report and review of literature. J Neurooncol. 2011; 101: 311-318.
- Lhermitte J, Duclos P. Sur un ganglioneuronme diffus du cortes du cervelet. Bulletin de l'Association française pour l'étude du cancer. 1920; 9: 99-107.
- Tan T-C, Ho L-C. Lhermitte-Duclos disease associated with Cowden syndrome. J Clin Neurosci. 2007; 14: 801-805.
- Kalyan Raman UP, Olivero WC. Ganglioglioma: a correlative clinicopathological and radiological study of ten surgically treated cases with follow-up. Neurosurgery. 1987; 20: 428-433.
- Luyken C, Blümcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J. Supratentorial gangliogliomas: Histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer. 2004; 101: 146-155.
- Chandrashekhar TN, Mahadevan A, Vani S, Yasha TC, Sampath S, Chandramouli BA, et al. Pathological spectrum of neuronal/glioneuronal tumors from a tertiary referral neurological Institute. Neuropathology. 2012; 32: 1-12.
- Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, VandenBerg SR. Ganglioglioma: an ultrastructural and immunohistochemical study. Cancer. 1997; 79: 989-1003.
- Lang FF, Epstein FJ, Ransohoff J, Allen JC, Wisoff J, Abbott IR, et al. Central nervous system gangliogliomas. J Neurosurg. 1993; 79: 867-873.
- Prayson RA, Khajavi K, Comair YG. Cortical architectural abnormalities and MIB1 immunoreactivity in gangliogliomas: a study of 60 patients with intracranial tumors. J Neuropathol Exp Neuro. 1995; 54: 513-520.
- Wolf HK, Mller MB, Spnle M, Zentner J, Schramm J, Wiestler OD. Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol. 1994; 88: 166-173.
- Wacker MR, Cogen PH, Etzell JE, Daneshvar L, Davis RL, Prados MD. Diffuse leptomeningeal involvement by a ganglioglioma in a child. J Neurosurg. 1992; 77: 302-306.
- Osborn A. Diagnostic Neuroradiology. St. Louis: Mosby; 1994.
- Grossman R, Yousem D. Neuroradiology. The Requisites. St. Louis: Mosby-Yearbook; 1994.
- Kikuchi T, Kumabe T, Higano S, Watanabe M, Tominaga T. Minimum apparent diffusion coefficient for the differential diagnosis of ganglioglioma. Neurol Res. 2009; 31: 1102-1107.
- Miller DC, Lang FF, Epstein FJ. Central nervous system gangliogliomas. J Neurosurg. 1993; 79: 859-866.
- Jay V, Squire J, Becker LE, Humphreys R. Malignant transformation in a ganglioglioma with anaplastic neuronal and astrocytic components. Report of a case with flow cytometric and cytogenetic analysis. Cancer. 1994; 73: 2862-2868.
- Kawataki T, Sato E, Sato T, Kinouchi H. Anaplastic Ganglioglioma with malignant features in both neuronal and glial components - A case report. Neurol Med Chir. 2010; 50: 228-231.
- Mittelbronn M, Schittenhelm J, Lemke D, Ritz R, Nägele T, Weller M, et al. Low grade ganglioglioma rapidly progressing to a WHO grade IV tumor showing malignant transformation in both astroglial and neuronal cell components. Neuropathology. 2007; 27: 463-467.
- Blümcke I, Müller S, Buslei R, Riederer BM, Wiestler OD. Microtubuleassociated protein-2 immunoreactivity: a useful tool in the differential diagnosis of low-grade neuroepithelial tumors. Acta Neuropathol. 2004; 108: 89-96.
- Yin X-L, Hui AB-Y, Pang JC-S, Poon WS, Ng H-K. Genome-wide survey for chromosomal imbalances in ganglioglioma using comparative genomic hybridization. Cancer Genet Cytogenet. 2002; 134: 71-76.
- Bhattacharjee MB, Armstrong DD, Vogel H, Cooley LD. Cytogenetic analysis of 120 primary pediatric brain tumors and literature review. Cancer Genet Cytogenet. 1997; 97: 39-53.
- Squire JA, Arab S, Marrano P, Bayani J, Karaskova J, Taylor M, et al. Molecular cytogenetic analysis of glial tumors using spectral karyotyping and comparative genomic hybridization. Mol Diagn. 2001; 6: 93-108.
- Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, et al. Activating mutations in BRAF characterize a spectrum of pediatric lowgrade gliomas. Neuro Oncol. 2010; 12: 621-630.
- Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011; 121: 397-405.
- Boer K, Troost D, Timmermans W, Van Rijen PC, Spliet WGM, Aronica E. Pi3K-mTOR Signaling and AMOG expression in epilepsy-associated glioneuronal tumors. Brain pathol. 2010; 20: 234-244.
- von Deimling A, Fimmers R, Schmidt MC, Bender B, Fassbender F, Nagel J, et al. Comprehensive allelotype and genetic anaysis of 466 human nervous system tumors. J Neuropathol Exp Neurol. 2000; 59: 544-558.
- Horbinski C, Kofler J, Yeaney G, Camelo-Piragua S, Venneti S, Louis DN, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain pathol. 2011; 21: 564-574.
- Hayashi Y, Iwato M, Hasegawa M, Tachibana O, von Deimling A, Yamashita J. Malignant transformation of a gangliocytoma/ganglioglioma into a glioblastoma multiforme: a molecular genetic analysis. J Neurosurg. 2001; 95: 138-142.
- Grajkowska W, Piekutowska-Abramczuk D, Ciara E, DembowskaBaginska B, Perek D, Roszkowski M, et al. Ganglioglioma associated with alterations of NBN gene. A case report. Folia Neuropathol. 2009; 47: 278- 283.
- Majores M, Schick V, Engels G, Fassunke J, Elger CE, Schramm J, et al. Mutational and immunohistochemical analysis of ezrin-, radixin-, moesin (ERM) molecules in epilepsy-associated glioneuronal lesions. Acta Neuropathol. 2005; 110: 537-546.
- Becker AJ, Löbach M, Klein H, Normann S, Nöthen MM, von Deimling A, et al. Mutational analysis of TSC1 and TSC2 genes in gangliogliomas. Neuropathol Applied Neurobiol. 2001; 27: 105-114.
- Kam R, Chen J, Blümcke I, Normann S, Fassunke J, Elger CE, et al. The reelin pathway components disabled-1 and p35 in gangliogliomas – a mutation and expression analysis. Neuropathol Appl Neurobiol. 2004; 30: 225-232.
- Parry L, Maynard J, Patel A, Hodges AK, von Deimling A, Sampson JR, et al. Molecular analysis of the TSC1 and TSC2 tumour suppressor genes in sporadic glial and glioneuronal tumours. Human Genetics. 2000; 107: 350-356.
- De Tommasi A, Luzzi S, D'Urso PI, De Tommasi C, Resta N, Ciappetta P. Molecular genetic analysis in a case of ganglioglioma: identification of a new mutation. Neurosurgery. 2008; 63: 976-980.
- Resta N, Lauriola L, Puca A, Susca FC, Albanese A, Sabatino G, et al. Ganglioglioma arising in a Peutz-Jeghers patient: a case report with molecular implications. Acta Neuropathol. 2006; 112: 106-111.
- Sawin PD, Theodore N, Rekate HL. Spinal cord ganglioglioma in a child with neurofibromatosis type 2. Case report and literature review. J Neurosurg. 1999; 90: 231-233.
- Naydenov E, Tzekov C, Minkin K, Nachev S. Malignant progression of anaplastic supratentorial ganglioglioma into glioblastoma multiforme in a patient with turner syndrome. J Neurol Surg A Cent Eur Neurosug. 2012; 73: 253-255.
- Cheung HC, Yatsenko SA, Kadapakkam M, Legay H, Su J, Lupski JR, et al. Constitutional tandem duplication of 9q34 that truncates EHMT1 in a child with ganglioglioma. Pediatr Blood Cancer. 2012; 58: 801-805.
- Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G, Foulds N, et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet. 2009; 46: 598-606.
- Yatsenko SA, Brundage EK, Roney EK, Cheung SW, Chinault AC, Lupski JR. Molecular mechanisms for subtelomeric rearrangements associated with the 9q34.3 microdeletion syndrome. Hum Mol Genet. 2009; 18: 1924- 1936.
- Zhou X-P, Marsh DJ, Morrison CD, Chaudhury AR, Maxwell M, Reifenberger G, et al. Germline Inactivation of PTEN and Dysregulation of the Phosphoinositol-3-Kinase/Akt Pathway Cause Human LhermitteDuclos Disease in Adults. Am J Hum Genet. 2003; 73: 1191-1198.
- Aronica E, Boer K, Becker A, Redeker S, Spliet WG, van Rijen PC, et al. Gene expression profile analysis of epilepsy-associated gangliogliomas. Neuroscience. 2008; 151: 272-292.
- Khashab ME, Gargan L, Margraf L, Koral K, Nejat F, Swift D, et al. Predictors of tumor progression among children with gangliogliomas. J Neurosurg Pediatr. 2009; 3: 461-466.
- Majores M, von Lehe M, Fassunke J, Schramm J, Becker AJ, Simon M. Tumor recurrence and malignant progression of gangliogliomas. Cancer. 2008; 113: 3355-3363.
- Selvanathan S, Hammouche S, Salminen H, Jenkinson M. Outcome and prognostic features in anaplastic ganglioglioma: analysis of cases from the SEER database. J Neurooncol. 2011; 105: 539-545.
- Luyken C, Blümcke I, Fimmers R, Urbach H, Elger CE, Wiestler OD, et al. The Spectrum of Long-term Epilepsy–associated Tumors: long-term seizure and tumor outcome and neurosurgical aspects. Epilepsia. 2003; 44: 822-830.
- Southwell DG, Garcia PA, Berger MS, Barbaro NM, Chang EF. Long-term seizure control outcomes after resection of gangliogliomas. Neurosurgery. 2012; 70: 1406-1414.
- Rades D, Zwick L, Leppert J, Bonsanto MM, Tronnier V, Dunst J, et al. The role of postoperative radiotherapy for the treatment of gangliogliomas. Cancer. 2010; 116: 432-442.
- Lee C-C, Wang W-H, Lin C-F, Chen H-H, Chen S-C, Lin S-C, et al. Malignant transformation of supratentorial ganglioglioma. Clinical Neurology and Neurosurg. 2012; 114: 1338-1342.