Research Article
Proteomics Tools – An Update
Sachin Kumar1*, Vikar Gaur, Sartaj Khurana2, Sudeep Bose3, Manjula Kiran4, Manjula Kiran5 and Surender K Sharawat6
1Department of Medical Oncology, Dr. B. R. Ambedkar Institute Rotary Cancer Hospital, India
2Department of Medical Oncology, Dr. B. R. Ambedkar Institute Rotary Cancer Hospital, India 3Department of Biotechnology, Amity University, Noida, Uttar Pradesh -201303, India
4Department of Biotechnology, Amity University, Noida, Uttar Pradesh -201303, India
5Deprtment of Biologicals, Amity University, India
6Department of Medical Oncology, Dr. B. R. Ambedkar Institute Rotary Cancer Hospital, All India Institute of Medical Sciences, India
*Corresponding author: Sachin Kumar, Department of Medical Oncology, Dr. B. R. Ambedkar Institute Rotary Cancer Hospital, India
Published: 10 Oct, 2017
Cite this article as: Kumar S, Gaur V, Khurana S, Bose
S, Kiran M, Kiran M, et al. Proteomics
Tools – An Update. Clin Oncol. 2017;
2: 1358.
Abstract
Recent advances in high-throughput genome sequencing, analytical instrumentation, bioinformatics and computational biology has permanently changed the way biology is practiced. With the evolution and progress in system biology, scientists can now study the systems comprehensively and in very fine molecular details rather than just focusing on various components of living system individually. Proteomics, commonly described as expressed form of genome, has emerged as one of the vital constituent of system biology and is being studied extensively across the globe. Unlike genome, proteome is a very dynamic and complex entity involving not only the structures, sequences, and functions of proteins, but also their abundance, localization, modifications, and interactions. Hence, proteomics can provide plenty of information for better understanding of the diversity and intricacies of the biological system. However, the analysis of these various properties of the proteome requires an equally diverse range of technologies, like two-dimensional gel electrophoresis, mass spectrometry, and protein microarrays. In this review, we provide an introduction to the field of proteomics with an aim to describe numerous tools and techniques to study proteomics.
Introduction
The term “proteome” was first used by Marc Wilkins, an Australian postdoctoral fellow, in 1994 [1]. He defined proteome as the entire set of proteins expressed in a specific cell at a specific time and the study of which is now known as “proteomics.” The term proteome is a combination of proteins and genome. Proteomics aims to study the dynamic protein products of the genome, including its structure, function, regulation and interactions, rather than focusing on the simple static DNA blueprint of a cell. In eukaryotes, the proteome is bigger than the genome, mainly due to alternative splicing of genes and various post-translational modifications (PTMs) a protein undergoes, like acetylation, phosphorylation, and glycosylation. As the field of proteomics grew further, it was soon realized that a specific protein we examine can result because of transcripts from identical genes, identical parts of genes or from various PTMs. This led to the concept of protein species which was defined as the smallest unit of the proteome [2]. Soon, it was evident that in proteomics, analysis of protein species can’t be ignored to get a comprehensive picture of the biological complexity. In addition to various post-translational protein modifications, there are different external and internal factors which affect protein expression at a particular time. Keeping all this in mind, proteome of an individual was defined as the sum of all the protein species which are expressed in a time-dependent manner during the life span of an organism [3]. Further, total set of protein species expressed in a specific biological compartment at a given time and under specific environmental situations is known as ‘sub-proteome’ [3]. One of the major challenges of the current proteomics technologies is to measure the entire set of proteins and protein species expressed in an individual from birth to death. Proteomics has been rightly called the ‘science in preparation for the new millennium’, due to rapid advances achieved in its automation, combinatorial chemistry and high throughput screening [4,5]. On account of its enormous potential, proteomics can be further divided into three branches: ‘structural proteomics’, ‘expression proteomics’ and ‘interaction proteomics’. All these branches of proteomics possess enormous potentials. Through structural proteomics, analysis of protein structure and its comparison can help in elucidation of the functions of newly identified genes. Structural proteomics can also be used to investigate drug-protein interactions and protein- protein interactions. In contrast, through expression proteomics, novel, disease-specific or differentially expressed proteins can be identified by comparing the expression levels of proteins in normal healthy tissues and diseased tissues and such proteins can represent a therapeutic target or can serve as diagnostic biomarkers. Determination of protein-protein interactions by interaction proteomics helps to determine protein functions and can also show how proteins assemble in larger complexes.
Figure 1
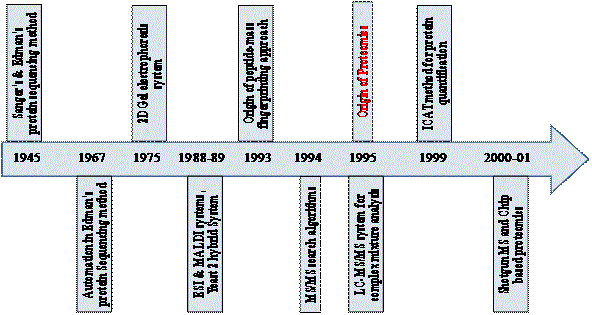
Figure 1
Breakthrough achievements along the time line leading to the origin of proteomics.
Figure 2
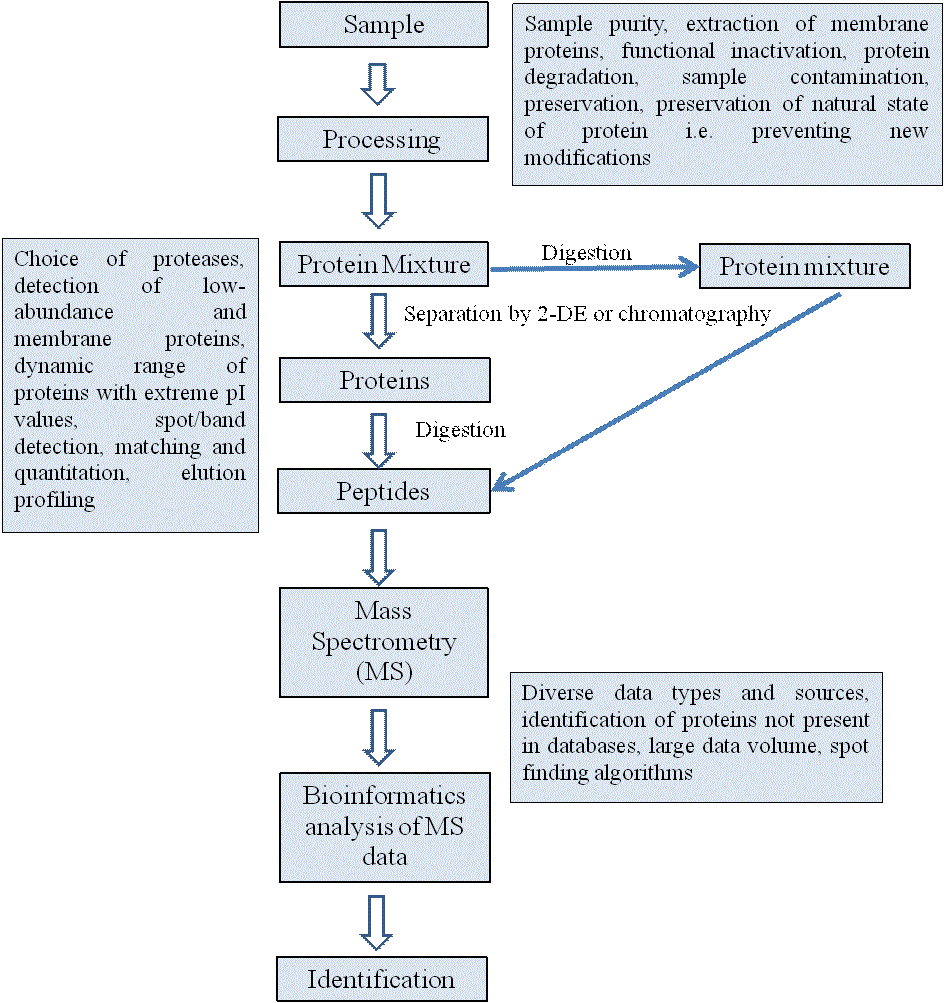
Figure 2
General flow scheme for proteomic analysis with highlighted considerations.
History
Most advances in biology can usually be traced back to the
development of new techniques. The field of proteomics is not an
exception. In 1945, the pioneering work by Frederick Sanger on
separation technology paved the way for the development of protein
sequencing [6]. However, protein sequencing gained momentum
only when the phenylisothiocyanate sequencing chemistry developed
by Edman in 1949 [7] was automated [8]. This was followed by
the development of first computer programs for the amino acid
sequencing [9,10]. The idea of analyzing the complete complement
of proteins being produced by a cell arose nearly four decades ago
with the development of two-dimensional gel electrophoresis (2-DE).
Margolis and Kenrick [11] used a combination of native isoelectric
focusing with pore gradient sodium dodecyl sulphate polyacrylamide
gel electrophoresis (SDS-PAGE) to separate serum proteins. The
2-DE technique which is mostly used today originated from the
work of Patrick O'Farrell [12] and Joachim Klose [13]. Several gel
analysis and database programs were developed such as PDQuest
[14], GELLAB [15], TYCHO (later KEPLER) [16], and MELANIE
[17]. This development in the field of 2-DE led to an idea of building
protein database such as human protein index [18]. In the late
1980s, inventions in the field of ionization techniques for peptides
and proteins: electrospray ionization (ESI) [19] and matrix-assisted
laser desorption/ionization (MALDI) [20,21] revolutionized the
field of mass spectroscopy (MS). The success obtained with MALDIand
ESI-based ionization techniques enabled the improvement of
commercial mass spectrometers by incorporation of these robust
‘ion source’, making them more acceptable methods for protein
identification amongst a variety of other proteomics technologies.
The field of proteomics grew further with the introduction of peptide
mass fingerprinting (PMF) in 1993 by several research groups [22-
26]. Developments in the field of MS led to the first published MS/
MS search algorithms [27,28] which further facilitated MS mediated
protein identification. The method of non-spray ESI was developed
subsequently [29,30] which was preferred for the chromatography
based MS approaches like LC-MS/MS.
With the rapid growth in analytical technologies and development
of various tools and databases in computational biology and
bioinformatics, it became feasible to extract, interrogate and identify
specific protein from complex protein mixtures with precision. With
this, in the year 1994, the term ‘proteome’ was coined in the firstever
2-DE meeting held in Siena, Italy [1]. Since then, the proteome
is popularly defined as the protein complement of the genome, while
its study is known as ‘proteomics’. Today’s proteomic practitioners
enjoy the legacy of the past 30 years (Figure 1).
Proteomic Tools
Proteomics is a complement to various genomics-based
approaches. One of the major advantages of proteomics is that the
protein is the endpoint of biological system. A cell gets it unique identity
because of a variety of proteins expressed in that cell type. Study of
protein dynamics, like changes in protein expression, modifications,
and interactions in a cell type in response to environmental conditions
and in pathogenic conditions, such as cancer, and also its intrinsic
genetic programs can help in better understanding of molecular
mechanisms involved in these processes. Expression and function of
proteins is regulated through transcription as well as PTMs. Different
splicing can yield more than one RNA from one gene. Moreover,
proteins can undergo >200 PTMs which can affect their function,
their interactions with other proteins and nucleic acids, trafficking,
stability, turn-over rate and so on [31], and this being contributing
to production of a large number of protein products from one gene.
Elucidating and understanding these changes at molecular levels are
the underlying themes in the proteomics.
The essential elements involved in analytical proteomics are
illustrated in figure 2. Briefly, a complex protein mixture consisting
of proteins of different molecular mass, solubilities and modifications
is separated into rather less complex mixtures consisting of fewer
components. This is usually achieved by the separation of intact
proteins first followed by their digestion into peptides or vice versa.
The separation is done by either 2-dimensional gel electrophoresis (2-
DE) or various chromatography-based approaches. The peptides are
analyzed by MALDI-TOF-MS or ESI-MS. The data generated by MS
is then matched with available databases using various bioinformatics
software for identifying peptides and their sequences. In the end,
identify of a protein is established from the original complex protein
mixture. During the last decade, new approaches have been developed
not only for the identification of various proteins expressed in cells but
also for comparing the expression levels of proteins under different
cellular context. In the following section, various classical as well as
emerging proteomics technologies are being described.
Two-Dimensional Gel Electrophoresis (2-DE)
2-DE has been the one of the most widely accepted technique
in quantitative proteomics [32]. 2-DE involves extraction of total
proteins from a given sample and their separation according to
isoelectric points (pIs) followed by molecular weights in first and
second dimension, respectively to produce protein profiles. The
reproducibility and resolution in separating almost entire range
of proteins (basic or acidic) has been enhanced significantly by
employing immobilized pH gradients in 2-DE [32,33]. By using large
format gels (40-30 cm as opposed to the ‘standard’ 20-20 cm format),
10,000 proteins have been resolved from mouse testis extracts [34].
Automated versions of 2-DE have also been established, e.g., 2-D
membrane electrophoresis, which provides a quick, high resolution
two-dimensional technique for protein separation on polyvinylidene
fluoride (PVDF) or nylon membranes without cooling effect. The
separation in both dimensions is performed on the same membrane,
which just has to be rotated in the electric field after the buffer change.
The whole process takes only 20 min as compared with 1-2 days for
conventional 2-DE polyacrylamide gel electrophoresis (PAGE).
The detection of different protein spots on the gel is achieved
using a variety of dyes and stains available in the market [35].
Coomassie blue dyes, silver stain, and fluorescent dyes are most
commonly used dyes in 2-DE. Autoradiography is also being used
to visualize and quantitate radiolabelled proteins that are resolved by
2-DE. Proteins are commonly labeled with either 3H, 14C, 35S, 32P or
125I in vivo [36]. Imaging of the stained gels is usually performed by
using charge-coupled device (CDD) camera, phosphor imagers and
multichannel array detectors. The obtained gel image is processed
using Photoshop or Corel programmes before applying an analysis
system. The quantification and identification of specific protein
resolved on a 2-DE gel can be achieved by scanning of stained gels
by laser densitometers set at different resolutions and with the use
of bioinformatics software like PDQuest [14]. One of the most
widely accepted applications of 2-DE has been to detect differential
protein expression in diseased versus healthy tissues. This has led to
identification of a number of protein-based biomarkers in various
human cancers.
Two-dimensional Differential Gel Electrophoresis (2D-DIGE)
2-DE has some serious limitations such as it is a time consuming and suffers from gel-gel variations. 2D-DIGE can be used to overcome these limitations as it depends on multiple fluorescent dyes, like Cy dyes, for the labeling of different protein samples followed by their separation and visualization on a single 2-DE gel [37]. Up to three protein samples can be labeled, each with a different fluorescent dye, (like Cy2, Cy3, and Cy5 dyes), then combined and separated by 2-D PAGE on a single gel. Densitometric scanning at different wavelengths, characteristic for each dye, permit to acquire three images from only one gel. Even small differences in expression levels can be determined by comparing the ratio obtained from one fluorescent-labeled sample directly with other using different wavelengths for image analysis. Both, the classical 2-DE as well as 2D-DIGE requires image analysis software such as Image Master 2D Platinum (GE Healthcare, Uppsala, Sweden) for classical gels and De Cyder (DeCyder Differential Analysis software, GE Healthcare) with regard to the DIGE gels.
Mass Spectrometry (MS)
Mass spectrometers are usually divided into three fundamental
parts, namely the ionization source, the analyzer, and the detector.
MS is a powerful tool to obtain peptide mass fingerprints (PMF)
for proteins which are resolved by 2-DE. In a typical proteomics
experiment, proteins resolved by 2-DE or DIGE are first subjected
to ‘in-gel’ digestion followed by extraction of peptides from the
gels and their ionization by feeding them into mass spectrometers.
The characterization of proteins has been greatly improved by
the availability of MALDI and ESI-based ionization techniques in
MS [19,20]. In MALDI, peptides are co-precipitated with a lightabsorbing
matrix and the mixture is exposed to short pulses of UV
light radiation under low pressure. As a result of this, peptides are
ionized and accelerated in an electric field followed by their movement
back through an energy-corrected machine [38]. Elapsed time is
measured from acceleration to-field-free drift by time-of-flight or by
a quadrupole detector from which mass of peptides can be derived.
A spectrum consisting of molecular mass of individual peptides
is generated, which is further used to search available databases to
establish the identity of proteins. Unlike MALDI, in ESI, peptides
are ionized by passing them through a capillary device at very high
voltage [38]. This makes peptides charged which are then directed
to a MS under very low pressure to separate them according to their
mass to charge ratios. Nano-electrospray ionization tandem MS,
commonly known as ESI MS/MS is another variant of MS approach
[29,30]. In ESI MS/MS, a microcapillary tube containing 1 ml of
peptide solution sprays a fine mist of charged droplets generated from
a potential difference between the capillary and the inlet to the mass
spectrometer. Desolvated peptides ions are formed as the solvent
evaporates in a high vacuum chamber, and are resolved to produce
the first MS scan. This is followed by selective targeting of peptide ion
in a collision chamber and its fragmentation after its interaction with
an inert gas. The fragments of the peptide ion are then resolved based
on their m/z ratio to generate the second MS spectrum with a series of
small peptides that differ only by a single amino acid. One of the major
advantages of ESI MS/MS is their capability to accurately identify
PTMs by incorporating the measurement of mass shifts. For the
separation of complex peptide mixtures, gel-independent methods like
liquid chromatography coupled with MS (LC-MS/MS) systems, has
also been developed [39]. One of such approach is known as ‘shotgun
proteomics’, in which LC-MS/MS and various protein sequence
databases are used together to study complex mixture of peptides
generated through proteolytic digestion of proteins isolated from a
sample [40,41]. This approach uses reversed-phase LC to separate the
tryptic digests of entire proteins followed by online ESI tandem MS
for peptide sequencing. The generated MS spectra of whole cellular
fraction are carefully analyzed and protein identification is performed
through peptide assignment and database searching. MALDI-TOF
MS has been widely used to identify differentially expressed proteins
in various human malignancies, like renal cell carcinoma [42] and
breast carcinoma [43]. Fourier transform ion cyclotron resonance
mass spectrometer (FTICR-MS) is another system developed to
identify proteins present in low abundance and to separate proteins
with closely related mass to charge ratios in complex mixtures [44].
A combination of FTICR-MS, HPLC and ESI were not only able
to differentiate single compounds present in big combinatorial
chemistry libraries but also to precisely identify peptide mass with
high-throughput mode. Imaging MS has been developed to identify
and localize biomolecules, like proteins, directly in tissue sections. In
imaging MS, a metal plate, which contains individual cells or frozen
tissue sections coated with UV-absorbing matrix, is placed in the
MS system and images are produced, depending upon selected mass
values, through an optical scanner which measures peak intensities in
tissue specimen over thousands of spots [45]. Imaging MS has great
potential in discovery of biomarkers, identifying aberrant localization
of biomarkers in normal and diseased conditions, understanding
molecular heterogeneity of cancer cells, and also in intra-operative
evaluation of surgical margins of tumours. Imaging MS has been
employed to discover biomarkers in glioblastomas [46].
Selected reaction monitoring (SRM)-MS is an emerging targeted
MS-based method specifically developed to complement untargeted
shotgun methods [47]. The major application of SRM is in the
measurements of a set of pre-selected protein-based biomarkers in
multiple samples. In such context, SRM has proven to yield consistent
and reproducible results with very high precision [48]. It uses
sensitive and specific mass spectrometric assays to measure target
analytes across multiple samples. Typically, in a single LC-MS test, a
large number of peptides are measured [49]. SRM involves selection
of a peptide ion and a fragment ion with predefined mass to charge
ratio by employing first and third quadrupoles as filters, whereas
collision occurs in second quadrupole. The major application of
SRM is in cancer research where it has been widely used for the
identification and measurements of wild type and mutant forms of
various proteins in cancer cells and bio-fluids as well as validation of
cancer biomarkers in a fast and cost-effective manner [50]. Due to
the increased scan speed and mass window selectivity of the current
mass analyzers, SRM can be simultaneously performed on multiple
analytes. This capability lead to the multiplexing of SRMs in a method
called multiple reaction monitoring (MRM). A number of high
resolution MS systems, such as QTOF and Orbitraps have employed
triple quadrupole for improved sensitivity, specificity and scan speed.
ABI5600 and Xevo G2 XS QTOF are newer MS systems which can
perform protein identification was well as their quantification. When
performed on QTOFs or Orbitraps, MSM is popularly called as high
resolution MRM (HR-MRM) or parallel reaction monitoring (PRM).
However, In PRM a particular fragment ion can’t be monitored
during acquisition. Generally, quantitation of selected fragment
ion is achieved by analyzing chromatogram generated after the
acquisition of mass spectrum. Because of fast scanning, it became
possible to develop sequential window acquisition of all theoretical
mass spectra (SWATH). In this method, information-dependent
acquisition i.e., IDA is used for creation of a spectral library and
system is set for selecting precursor ions using a mass range (400-
1250 m/z). Finally, the required SWATH-MS data is extracted for
the quantification of peptides. MALDI-Imaging (MALDI-MSI) is
an emerging tool to investigate the molecular details of tissues in
their morphological context and is also being seen as a substitute for
immunohistochemistry [51]. It allows direct quantification of various
biomolecules, such as proteins, peptides, and metabolites from tissue
specimens. An image is captured which is specifically related with the
distribution of detected biomolecule. As MALDI-MSI doesn’t involve
use of radiolabeled or fluorescently labeled reagents, it is rapidly
being accepted for its use in tissue-based biomarker discovery.
Accessible body fluids such as blood, urine or saliva can be then used
for verification of the presence of these specific markers in the aim
of developing non-invasive tests for cancer diagnosis or screening.
When combined with 3D reconstruction of tissue composition and
other imaging tools like PET, CT scan, and MRI, MALDI-MSI can
help in the development of profile signature diagnosis for early
detection of cancer and to complement histopathology, assessment of
therapeutic efficacy and personalized medicine [52,53].
Liquid Chromatography and Multi-Dimensional Separation Technology
Liquid chromatography (LC) based separation methods are common in proteomics because of their versatility and compatibility with mass spectrometry. LC can be applied either upstream of 2-DE to pre-fractionate the sample (Figure 3), downstream of 2-DE to separate the peptide mixtures from single excised spots, or instead of 2-DE as the major protein separation technology. Alternative LC methods exploiting different separation principles, such as size, charge, hydrophobicity and affinity for particular ligand, are also being used in proteomics. As is the case for electrophoresis, the highest-resolution separations are achieved when two or more separation principles are applied one after the other in orthogonal dimensions.
Figure 3
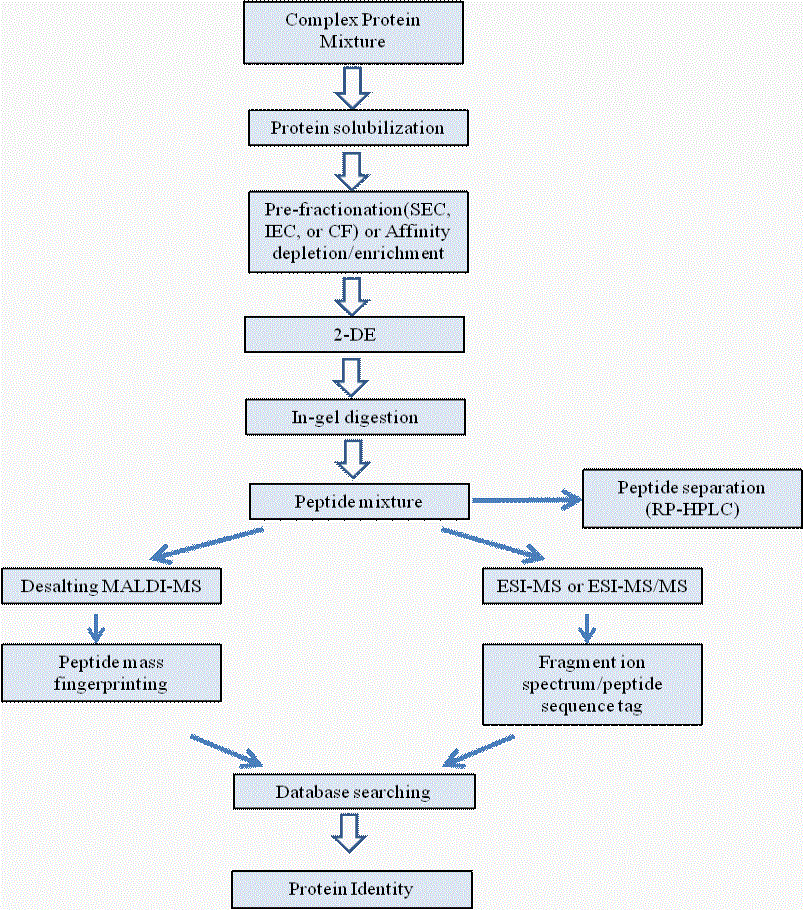
Figure 3
Liquid chromatography in combination with 2-DE for standard proteomic analysis.
Analysis
Abbreviation: SEC, size exclusion chromatography; IEC, ion exchange chromatography; CF, chromatofocusing; 2-DE, 2-dimensional gel electrophoresis; RP-HPLC, reverse-phase HPLC. Apart from LC and 2-DE based technologies, other alternative chromatography techniques are also being used for proteomic studies [54]. Employed a biphasic two-dimensional micro (μ) liquid chromatography (LC) column packed with strong cation exchange and reversed phase materials for step-wise analysis of peptides initially by charge followed by hydrophobicity. The multi-dimensional protein identification technology (MuDPIT; Figure 4) system was built-in-line with a tandem mass spectrometer. With this approach protein of high (>100 kDa)/low (<10kDa) mass and of relatively low abundance (codon bias <0.2) and solubility could be identified; thereby extending the analysis range possible because proteins and peptides are separated by different physicochemical properties than with 2-DE. Multi-dimensional approaches can combine sizeexclusion [55], ion-exchange [56], immobilized metal affinity [57], reversed-phase chromatography or capillary electrophoresis [58] and are either directly integrated with an MS instrument or run off-line prior to MS identification [59].
Peptide Separation
Abbreviation: SEC, size exclusion chromatography; IEC, ion exchange chromatography; CF, chromatofocusing; 2-DE, 2-dimensional gel electrophoresis; RP-HPLC, reverse-phase HPLC; CE, micro-capillary electrophoresis.
Surface-Enhanced Laser Desorption/Ionization (SELDI)
SELDI is based on a patented method for the separation, profiling, and analyzing proteins which are expressed at femtomole levels [60]. In SELDI, specific probe surfaces or chips are used for the affinitybased capture of proteins. The most critical component of SELDI is the chips, which usually have broad binding properties based on immobilized metal affinity capture or biochemically modified surfaces like antibodies and receptors [61]. After their capture on the protein chip array, proteins are detected by MS system where individual proteins are presented as separate peaks based on their mass to charge ratio and a retentate map is generated. Ciphergen’s ProteinChip assay SELDI-MS system is one of the popular commercially available platform based solely on protein biochip [61]. This system has been used to improve the diagnostic efficacy of low-grade bladder cancer to 75% as compared to the 30% when detected with urine cytology [62]. This system has also been used in identifying protein biomarkers expressed by prostate cancer, lung cancer, ovarian cancer and breast cancer.
Figure 4
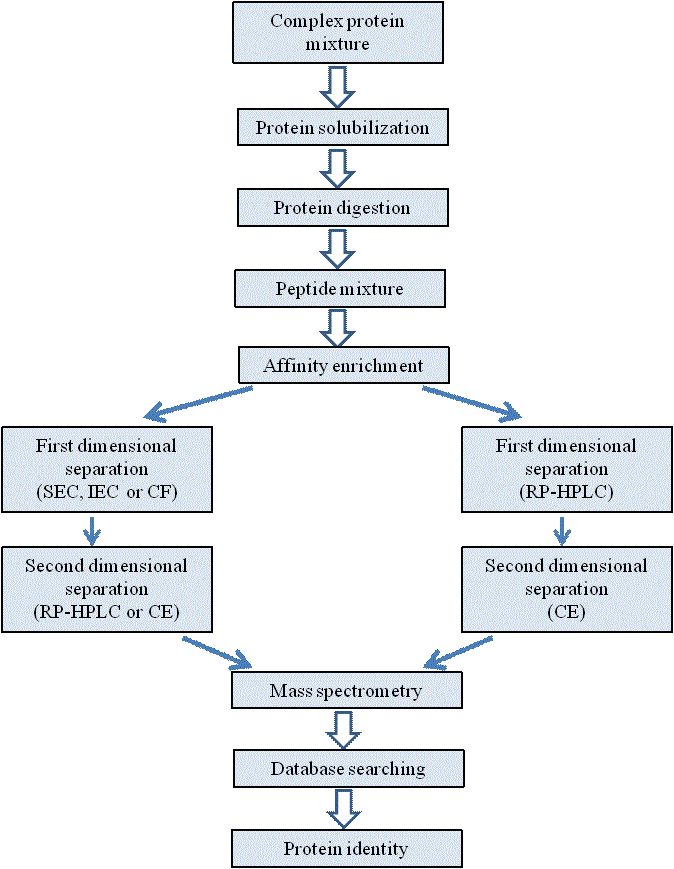
Figure 4
A scheme of multi-dimensional protein identification technology (MuDPIT) for peptide separation.
Proteins and Antibody Microarrays
Protein microarray, also known as protein chips, are one of the most promising high throughput techniques developed for the characterization of proteins [63]. A basic protein microarray consists of an array of immobilized protein spots filled with ‘bait’ proteins which are hybridized with small molecular probes labeled with fluorescent tags. There are currently three variants of protein microarrays that are used to study the biochemical activities of the proteins namely, (i) analytical microarrays, (ii) functional microarrays, and (iii) reverse phase microarrays. Analytical microarrays are used widely for monitoring protein expression levels, biomarker identification, clinical diagnosis etc. On the contrary, functional microarrays comprise of whole protein domains and are employed for probing of many protein activities and protein interactions. Reverse phase microarray is used to determine the presence of modified disease causing proteins. For instance, post-translational modifications can be detected using reverse phase microarrays [64]. Of all the analytical protein microarrays, the most widely used is the antibody array. It is an ELISA-based high throughput technique that is used for protein characterization. This technique enunciates the detection of proteins post antibody capture using direct protein labeling. Antibody arrays have two main variants: (i) labeled based assays and (ii) sandwich assays. The labeled based assay involves the labeling of target proteins with a tag allowing detection after the immobilized antibody is captured. This assay can allow incubation of two different samples at the same time- a reference and a test sample each labeled with a different tag to avoid the variation that may arise between the spots after antibody capture. There are drawbacks to this assay too such as limited specificity and sensitivity. The other variant, sandwich assay, involves the capture of unlabeled proteins by immobilized antibodies and these captured proteins are detected by a second antibody. These assays have higher sensitivity and specificity as compared to the labeled based assays [65]. Protein arrays are now being used extensively for characterization of cancer biomarkers from samples ranging from serum to tissue culture lysates to tumor biopsies and have proved themselves to be an essential tool in cancer research. For the identification and tracking of progression of cancer, immobilized proteins derived from purified cancer cells of micro dissected cells have been used as protein arrays [66]. Antibody arrays have also been used for the discovery of differentially expressed proteins in cancer versus non-cancerous tissue for their use as cancer biomarkers. Recently, many antibody arrays for specific detection of different cytokines have been established. For example, TranSignalTM (Panomics, Inc. Frement, California) cytokine antibody array, where multiple cytokines can be measured in a single hybridization experiment. These arrays have detection sensitivity in the range of pg/ ml for cytokines and an array consistency of zero and 10% between the same spots on two similar membranes.
Isotope-Coded Affinity Tags (ICAT)
Isotope-coded affinity tags depends upon the chemical labeling of any pair of protein samples with two identical reagents isotopic ally different in mass, allowing the relative amount of protein to be quantitatively compared in the subsequent mass spectral determination [67]. A typical ICAT reagent is made up of 3 components: i) an affinity tag, usually a biotin-based, for the isolation of ICAT-labeled peptides, ii) a linker for the incorporation of stable isotopes, and iii) a thiol specific reactive group. There are two varieties of ICAT reagent, light (without deuteriums) and heavy (with 8 deuteriums), with a difference in molecular weight of 8Da between them. A pair of protein samples are labeled on their cysteine residues, respectively, with either light or a heavy form of ICAT chemical reagents and then mixed together for proteolytic digestion. The digested mixture is purified through avidin affinity chromatography using biotin tags on ICAT reagents to isolate the ICAT labeled peptides. These peptides are subjected to MS analysis for production of peak ratios of various proteins which is followed by their sequencing through MS/ MS resulting in the identification of proteins expressed in abnormal levels. Thus, in a single automated process, identification and relative abundance of proteins in two related samples can be determined. Thus, the major application of ICAT is in discovery of protein-based diagnostic and prognostic biomarkers and therapeutic drug targets in various human disease conditions, including cancer. Variations of ICAT approach like, alternative labeling chemistries [68]; stable isotope labeling [69]; incorporation of stable isotopes of oxygen (16O or 18O) at the C-terminus of peptides [70,71]; and metabolic labeling of proteins by stable isotopes has also been attempted in a LC-MS/ MS system [72-74].
Isobaric Tag for Relative and Absolute Quantitation (Itraq)
iTRAQ is another MS-based approach for comparative measurements of single peptide or protein in a complex protein mixture [75]. Four iTRAQ reagents e.g., 114, 115, 116, and 117, which are chemically identical and have same molecular mass, can be used to multiplex four unrelated samples for relative quantification of proteins or peptides. A typical iTRAQ label is made up of a 145Da isobaric tag containing carbonyl balancer group and N-methylpiperazine-based reporter group and a NHS ester-based peptide reactive group [76]. The work-flow of iTRAQ experiment involves reduction and denaturation of protein samples, blocking of cysteine residues, and tryptic digestion of proteins followed by their labeling with iTRAQ reagents. This is followed by mixing equal amount of iTRAQ reagent-labeled samples into one sample mixture for LC-MS/MS analysis. Peptide sequence can then be produced from the product ions derived from the cleavage about peptide interresidue bonds. Relative abundance of specific peptides can then be checked in different samples by evaluating the intensities of reporter ion signals from MS/MS scan. As all the above steps are performed in a single tube, errors in protein identification and quantification which may result from sample loss in individual samples is avoided.
Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)
SILAC is an in-vivo process in which non radioactive label is
incorporated into proteins for MS analysis resulting in the detection
of relative abundance of proteins in different sample types. Amino
acids with substituted stable nuclei are incorporated into the protein.
This technique involves simultaneous culturing of two populations of
cells; one population is fed with growth medium containing a normal
amino acid, while the other cell population is fed with a medium
with heavy non-radioactive labeled amino acid. The cells with the
labeled amino acid in the medium incorporate this heavy isotope of
the amino acid into the proteins. All the proteins with the isotope
will be heavier than the normal counterparts. Using MS analysis, the
proteins can be segregated based on their mass difference [77].
SILAC technology is a powerful tool for identification of cancer
biomarkers. Proteins present in the amino acid labeled cells, could
prove to be effective chemotherapy targets. Recently, SILAC method
was followed to identify protein biomarkers indicative of resistance
to paclitaxel in lung cancer patients [78]. Tumor suppressor protein
programmed cell death protein 4 (PDCD4) was down regulated
by paclitaxel and elevated expression of PDCD4 was positively
correlated with longer survival of patients treated with paclitaxel [78].
In another study, two metastatic prostate cancer cell lines, PC3M
and PC3M-LN4 were used for SILAC followed by LC-MS/MS-based
proteomic approach. With this approach, E-cadherin was found to be
highly expressed in PC3M while UCHL1 was predominantly detected
in highly metastatic PC3M-LN4. Another protein found exclusively
in PC3M was Suppressor of Tumorigenecity-14, a protein expressed
in many normal epithelial tissues but absent in tumor lines derived
from these same sites [79]. Thus, SILAC can be effective method for
the identification of metastasis-related proteins which can be used for
drug designing and development and molecular targeted therapy.
Yeast Two-Hybrid System
This is a genetic proteomic assay developed for studying proteinprotein interactions [80]. It involves fusion of a DNA-binding domain with a protein of interest, i.e. bait and fusion of other proteins i.e. preys with a transcription-activating domain. Physical interactions between bait and prey are screened based on the activation of a transcription reporter construct. Yeast two-hybrid system has been widely used for screening of protein interactions in yeast [81], Helicobacter pylori [82], and Caenorhabditis elegans [83].
Affinity Tagging and Mass Spectroscopy
In yeast, affinity tagging and MS approach has been used for the efficient characterization of its protein complexes [84,85]. Briefly, endogenous genes of yeast and coding sequences of a tandem affinity purification tag are fused together. This is followed by purification and separation of tagged proteins along with their associated partners by gel electrophoresis and their identification by MS. Using this approach, Gavin et al purified and resolved 232 protein complexes containing 1440 different proteins in yeast [84]. Similarly, Ho et al identified > 3000 protein interactions consisting of 1578 individual proteins in yeast [85]. Affinity chromatography, fluorescence resonance energy transfer (FRET), and Surface Plasmon Resonance (SPR) are other methods used to identify protein-protein reactions.
Studying Protein Modifications
PTMs to protein structures play an important role in regulation of protein activity. Identification of the location and the type of modification usually give vital information for better understanding of the regulation and function of a specific protein in cellular or biological context. PTMs can be analyzed using various strategies. For example, phosphorylation can be detected by Western blotting using phosphorylated amino acids specific antibodies [86]. Tyrosine phosphorylated proteins have been detected by a combination of Western blotting after 2-DE and MS [87]. In contrast, enrichment of phosphorylated peptides either by immunoprecipitation or by metal-chelate affinity chromatography can be used to identify phosphoproteins [88]. Trypsin digestion and MS analysis is performed on these enriched phosphoproteins for the identification of resulting fragments. The concept of proteome analysis of glycoproteins from 2-D gels and analysis of model proteins has been proposed [89]. After separation of glycoprotein by 2-DE, protein spots are digested and glycopeptides identification and assignment of the glycosylation sites are carried out. A gel-free strategy was developed for the study of lectin-selected glycoproteins from trypsin digested sample of total protein [90]. To identify multiple PTMs in a single test, MacCoss et al employed shotgun MS approach and identified 270 proteins from human lens tissue [91]. Detailed analysis of lens crystallins proteins belonging to 11 families led to identification of a total of 73 PTMs. There have been attempts at a comprehensive analysis of modifications which include: ubiquitination [92], farnesylation [93] and glycosylphophatidylinositol anchoring of proteins [94].
Proteogenomics
Proteogenomics is an integrated approach employing a combination of proteomics, genomics and transcriptomics data to identify, assign and confirm the functional gene [95]. Such an approach requires handling of vast amount of information generated through different technologies employed in genomics (exome sequencing), transcriptomics (microarray, RNA-sequencing), and proteomics (LC-MS/MS). Since metabolomics i.e., the study of metabolites, also depends on the technologies similar to proteomics (LC-MS/MS), its integration in proteogenomics in inevitable in near future. In the work-flow of proteogenomics, a customized database of protein sequence is created which is entirely based on genomic information and the information obtained is utilized to analyze the model of interest. With the recent evolution in high-throughput sequencing platforms, theoretically, total set of protein-encoding sequences available in particular cells or tissues can be known. Thereafter, ‘shotgun proteomics’ can be used to identify proteins by searching the customized database of protein sequences. In this way, proteogenomics can prove to be very useful in the validation of gene expression at protein level [95]. Proteogenomics has huge potential in medical diagnostics and cancer research. Tumorspecific proteins or peptide signatures can be identifies using Oncoproteogenomics [96]. Such approach has recently been used to identify five protein signatures in The Cancer Genome Atlas (TCGA) cohort of colorectal cancer [97]. Amplification of chromosome 20q was found to be associated with significant global variations at mRNA as well as protein levels indicating it as a potential biomarker and a candidate therapeutic target [97]. In future, proteogenomics can also be used to classify cancers according to their molecular signatures and for the constitution of a personalized gene- or protein-based database.
Conclusion and Future Prospective
Proteomics is a field that promises to bridge the gap between genome sequence and cellular behavior. Initially, proteomics solely focused on studying the identity and measuring the expression levels of proteins. However, the development in the field of instrumentation, automation of large-scale analytical tools and emergence of various bioinformatics tools has shifted the focus to a broader area. This has allowed proteomic researchers to measure all of the biologically relevant properties of the proteins, including PTMs, interactions, folding etc. However, a significant challenge exists in the integration and automation of various proteomics technologies, a factor that underpinned the success of the large-scale DNA-sequencing projects. Important hurdles must be overcome at every stage of analysis, from sample preparation through to database management (Figure 2). Unlike DNA, proteins can’t be amplified and hence proteomics studies are restricted to proteins derived from natural sources making them substrate dependent. Another major challenge in the field of proteomics for the better understanding of any biological system is the extreme complexity associated with the proteome in terms of structure, stability, localization, function and dynamic range of abundance. The fact is that current technologies are simply inadequate for covering a whole proteome, even if statistics and sampling were not an issue. Although progress is being made on lowabundance- protein detection, we are nevertheless still far from even the possibility of producing a complete proteome for any organism for reasons of both complexity and dynamic range. Proteomic researchers will have to be more resourceful, especially as the tools used in today’s high-throughput environment still bear the stamp of an earlier era when one protein at a time was the standard. Clinical proteomics offers the opportunity and the potential to develop new diagnostic and prognostic tests, to identify new therapeutic targets, and eventually to allow the design of individualized patient treatment. With the advancements in proteomic technologies, disease-specific proteins have been identified for monitoring disease progression or therapeutic response. However, in clinical proteomics, sensitivity and specificity are two important determinants of the success of any protein marker. The future research will have to focus on improving techniques and technologies to achieve better sensitivity and specificity from the current proteomic tools. In this context, “technologies” refers to those instruments or instrumental approaches that provide fundamental capabilities, such as MS instruments, ion sources, chromatographic instrumentation, and so on. “Techniques,” on the other hand, are the procedures we use to get the most out of the available instrumentation. It is important to distinguish these two areas, because improvements in both will drive future progress in proteomics. Finally, it should be emphasized that proteomics is a young science, many of the technologies used in proteomics are still prototypical and that better materials, instrument design and methodology are expected to improve sensitivity, resolution and repeatability in the future.
References
- Wilkins MR, Sanchez JC, Gooley AA, Appel RD, Humphery-Smith I. Progress with proteome projects: why all proteins expressed by a genome should be identified and how to do it. Biotech Gen Eng Rev. 1995;13:19-50.
- Jungblut P, Thiede B, Zimny-Arndt U, Muller EC, Scheler C. Resolution power of two-dimensional electrophoresis and identification of proteins from gels. Electrophoresis. 1996;17:839-847.
- Jungblut PR, Holzhütter HG, Apweiler R, Schlüter H. The speciation of the proteome. Chemistry Central Journal. 2008;2:16-25.
- Tranter D. Preparing for the new millennium. Drug Discov Today. 1999;4:1-2.
- Blackstock WP, Weir MP. Proteomics: quantitative and physical mapping of cellular proteins. Trends Biotechnol. 1999;17:121-127.
- Sanger F. The free amino groups of insulin. Biochem J. 1945;39:507-512.
- Edman P, Begg G. A protein sequenator. Eur J Biochem. 1967;1:80-91.
- Edman P. Method for determination of the amino acid sequence in peptides. Acta Chem Scand. 1950; 4: 283–293.
- Kitagishi T, Hong YM, Shimonishi Y. Computer-aided sequencing of a protein from the masses of its constituent peptide fragments. Int J Pept Protein Res. 1981;17:436-443.
- Matsuo T, Matsuda H, Katakuse I. Computer program PAAS for the estimation of possible amino acid sequence of peptides. Biomed Mass Spectrom. 1981;8:137-143.
- Margolis J, Kenrick KG. 2-dimensional resolution of plasma proteins by combination of polyacrylamide disc and gradient gel electrophoresis. Nature. 1969;221:1056-1057.
- O’Farrel PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250: 4007-4021.
- Klose J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik. 1975;26:231-243.
- Garrels JI . The QUEST system for quantitative analysis of two-dimensional gels. J Biol Chem. 1989; 264: 5269-5282.
- Lemkin PF, Lipkin LE . Gellab: a computer system for two-dimensional gel electrophoresis analysis. III. Multiple two-dimensional gel analysis. Comp Biomed Res. 1981;14:407-446.
- Anderson NL, Taylor J, Scandora AE, Coult, BP, Anderson NG. The TYCHO system for computer analysis of two-dimensional gel electrophoresis patterns. Clin Chem. 1981;27:1807-1820.
- Appel RD, Hochstrasser DF, Funk M, Vargas JR, Pellegrini C. The melanie project: from a biopsy to automatic protein map interpretation by computer. Electrophoresis. 1991;12:722-735.
- Anderson NG, Anderson L. The human protein index. Clin Chem. 1982;28:739-748.
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989; 246: 64-71.
- Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 Da. Anal Chem. 1988;60:2299-2301.
- Tanaka K, Waki H, Ido Y, Akita S, Yoshida T. Protein and polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1988;2:151-153.
- Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C. Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases. Proc Nat Acad Sci USA. 1993;90:5011-5015.
- James P, Quadroni M, Carafoli E, Gonnet G. Protein identification by mass profile fingerprinting. Biochem Biophys Res Commun. 1993;195:58-64.
- Mann M, Hojrup P, Roepstorff P. Use of mass spectrometric molecular weight information to identify proteins in sequence databases. Biol Mass Spectrom. 1993;22:338-345
- Pappin D, Hojrup P, Bleasby A. Rapid identification of proteins by peptide-mass finger printing. Curr Biol. 1993;3:327-332.
- Yates JR III, Speicher S, Griffin PR, Hunkapiller T. Peptide mass maps: A highly informative approach to protein identification. Anal Biochem. 1993;214:397-408.
- Mann M, Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994;66:4390-4399.
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976-989.
- Wilm MS, Mann M. Electrospray and Taylor-Cone theory, Dole’s beam of macromolecules at last? Int J Mass Spectrom Ion Proc. 1994;136:167-180.
- Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature. 1996;379:466-469.
- Banks RE, Dunn MJ, Hochstrasser DF, Sanchez J-C, Blackstock W. Proteomics: new perspectives, new biomedical opportunities. Lancet. 2000; 356: 1749-1756.
- Hanash SM. Biomedical applications of two-dimensional electrophoresis using immobilized pH gradients: current status. Electrophoresis. 2000;21:102-109.
- Gorg A, Obermaier C, Boguth G, Harder A, Scheibe B. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis. 2000;21:1037-1053.
- Klose J, Kobalz U. Two-dimensional electrophoresis of proteins: an updated protocol and implications for a functional analysis of the genome. Electrophoresis. 1995;16:1034-1059.
- Patton WF. Detection technologies in proteome analysis. Journal of Chromatography B. 2002;771: 3-31.
- Link AJ. Autoradiography of 2-D gels. In: 2-D Proteome Analysis Protocols. 1999;285-290.
- Unlü M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis.1997;18:2071-2077.
- Andersen JS, Mann M (2000) Functional genomics by mass spectrometry. FEBS Letters.2 000;480:25-31.
- Appella E, Padian EA, Hunt DF (1995) Analysis of the structure of naturally processed peptides bound by class I and II major histocompatibility complex molecules. 1995;73:105-119.
- Spahr CS, Susin SA, Bures EJ, Robinson JH, Davis MT. Simplification of complex peptide mixtures for proteomic analysis: reversible biotinylation of cysteinyl peptides. Electrophoresis. 2000;21: 1635-1650.
- Wolters DA, Washburn MP, Yates JR III. An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73:5683-5690.
- Sarto C, Marocchi A, Sanchez JC, Giannone D, Frutiger S. Renal cell carcinoma and normal kidney protein expression. Electrophoresis. 1997;18:599-604.
- Vercoutter-Edouart AS, Lemoine J, Le Bourhis X, Louis H, Boilly B. Proteomic analysis reveals that 14-3-3s is down regulated in human breast cancer cells. Cancer Research. 2001;61:76-80.
- Schmid DG, Grosche P, Bandel H, Jung G. FTICR-mass spectrometry for high-resolution analysis in combinatorial chemistry. Biotechnol Bioeng. 2001;71:149-161.
- Chaurand P, Stoeckli M, Caprioli RM. Direct profiling of proteins in biological tissue sections by MALDI mass spectrometry. Annals of Chemistry. 1999;71:5263-5270.
- Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues. National Medicine. 2001;7:493-496.
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9:555-566.
- Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27:633-641.
- Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009;138:795-806.
- Selevsek N, Matondo M, Sanchez Carbayo M, Aebersold R, Domon B. Systematic quantification of peptides/proteins in urine using selected reaction monitoring. Proteomics. 2001;11:1135-1147.
- Chaurand P, Schwartz SA, Reyzer ML, Caprioli RM. Imaging mass spectrometry: principles and potentials. Toxicol Pathol. 2005;33:92-101.
- Franck J, Arafah K, Elayed M, Bonnel D, Vergara D. MALDI imaging mass spectrometry: state of the art technology in clinical proteomics. Mol Cell Proteomics. 2009;8:2023-2033.
- Andersson M, Groseclose MR, Deutch AY, Caprioli RM. Imaging mass spectrometry of proteins and peptides: 3D volume reconstruction. Nat Methods. 2008;5:101-108.
- Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676-682.
- Opiteck GJ, Jorgenson JW. Two-dimensional SEC/RPLC coupled to mass spectrometry for the analysis of peptides. Anal Chem. 1997;69:2283-2291.
- Davis MT, Beierle J, Bures ET, McGinley MD, Mort J. Automated LCLC-MS-MS platform using binary ion-exchange and gradient reversedphase chromatography for improved proteomic analyses. J Chromatogr B Biomed Sci Appl. 2001;752:281-291.
- Cao P, Stults JT. Mapping the phosphorylation sites of proteins using online immobilized metal affinity chromatography/capillary electrophoresis/ electrospray ionization multiple stage tandem mass spectrometry. Rapid Commun Mass Spectrom. 2000;14:1600-1606.
- Tong W, Link A, Eng JK, Yates JR. 3rd Identification of proteins in complexes by solid-phase microextraction/multistep elution/capillary electrophoresis/tandem mass spectrometry. Anal Chem. 1999; 71:2270- 2278.
- Figeys D, Gygi SP, Zhang Y, Watts J, Gu M. Electrophoresis combined with novel mass spectrometry techniques: powerful tools for the analysis of proteins and proteomes. Electrophoresis. 1998; 19:1811-1811.
- Jr GW, Cazares LH, Leung SM, Nasim S, Adam BL. Proteinchip(R) surface enhanced laser desorption/ionization (SELDI) mass spectrometry: a novel protein biochip technology for detection of prostate cancer biomarkers in complex protein mixtures. Prostate Cancer and Prostatic Disease. 1999;2: 264-276.
- Merchant M, Weinberger SR. Recent advancements in surfaceenhanced laser desorption/ionization time of flight-mass spectrometry. Electrophoresis. 2000;21:1164-1167.
- Vlahou A, Schellhammer PF, Mendrinos S, Patel K, Kondylis FI. Development of a novel proteomic approach for the detection of transitional cell carcinoma of the bladder in urine. American Journal of Pathology. 2001;158:1491-1502.
- Sutandy FX, Qian J, Chen CS, Zhu H. Overview of protein microarrays. Curr Protoc Protein Sci. Wiley Online Library.2013.
- Chen CS, Zhu H. Protein microarrays. BioTechniques. 2006;40:423-429.
- Haab BB. Antibody arrays in cancer research. Molecular & cellular proteomics. 2005;4:377-83.
- Paweletz CP, Charboneau L, Bichsel VE, Simone NL, Chen T. Reverse phase protein microarrays, which capture disease progression, show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20:1981-1989.
- Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH. Quantitative analysis of complex protein mixtures by isotope-coded affinity tags. Nature Biotechnology. 1999;17:994-999.
- Cagney G, Emili A. De novo peptide sequencing and quantitative profiling of complex protein mixtures using mass-coded abundance tagging. Nat Biotechnol. 2002;20:163-170.
- Zhou H, Ranish JA, Watts JD, Aebersold R. Quantitative proteome analysis by solid-phase isotope tagging and mass spectrometry. Nat Biotechnol. 2002;20:512-515.
- Mirgorodskaya OA, Kozmin YP, Titov MI, Körner R, Sönksen CP. Quantitation of peptides and proteins by matrix-assisted laser desorption/ ionization mass spectrometry using 18O labeled internal standards. Rapid Commun Mass Spectrom. 2000;14:1226-1232.
- Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836-2842.
- Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci USA. 1999;96:6591-6596.
- Conrads TP, Alving K, Veenstra TD, Belov ME, Anderson GA, et al. (2001) Quantitative analysis of bacterial and mammalian proteomes using a combination of cysteine affinity tags and 15N-metabolic labeling. Anal Chem. 2001;73:2132-2139.
- Smith RD, Pasa-Tolić L, Lipton MS, Jensen PK, Anderson GA. Rapid quantitative measurements of proteomes by Fourier transform ion cyclotron resonance mass spectrometry. Electrophoresis. 2001;22:1652- 1668.
- Wiese S, Reidegeld KA, Meyer HE, Warscheid B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics. 2007;3:340-350.
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Molecular & Cellular Proteomics. 2004;3:1154- 1169.
- Mann M. Functional and quantitative proteomics using SILAC. Nature reviews Molecular cell biology. 2006;7:952-958.
- Xu H, Dephoure N, Sun H, Zhang H, Fan F. Proteomic profiling of paclitaxel treated cells indentifies a novel mechanism of drug resistance mediated by PDCD4. J Proteome Res. 2015;14:2480-2491.
- Everley PA, Bakalarski CE, Elias JE, Waghorne CG, Beausoleil SA. Enhanced analysis of metastatic prostate cancer using stable isotopes and high mass accuracy instrumentation. J Proteome Res. 2006;5:1224-1231.
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340: 245-246.
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623-627.
- Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409:211-215.
- Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science. 2000;287:116-122.
- Gavin AC, Bösche M, Krause R, Grandi P, Marzioch M. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415: 141–147.
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180-183.
- Kaufmann H, Bailey JE, Fussenegger M. Use of antibodies for detection of phosphorylated proteins separated by two-dimensional gel electrophoresis. Proteomics. 2001;1:194-199.
- Yanagida M, Miura Y, Yagasaki K, Taoka M, Isobe T. Matrix assisted laser desorption/ionization-time of flight-mass spectrometry analysis of proteins detected by anti-phosphotyrosine antibody on two-dimensionalgels of fibrolast cell lysates after tumor necrosis factor-alpha stimulation. Electrophoresis. 2000;21:1890-1898.
- Griffin TJ, Aebersold R. Advances in proteome analysis by mass spectrometry. J Biol Chem. 2001;276:45497-45500.
- Packer NH, Pawlak A, Kett WC, Gooley AA, Redmond JW. Proteome analysis of glycoforms: a review of strategies for the microcharacterisation of glycoproteins separated by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis. 1997;18:452-460.
- Geng M, Zhang X, Bina M, Regnier F. Proteomics of glycoproteins based on affinity selection of glycopeptides from tryptic digests. J Chromatogr B. 2001;752:293-306.
- MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci USA. 2002;99:7900–7905.
- Layfield R, Tooth D, Landon M, Dawson S, Mayer J. Purification of polyubiquitinated proteins by S5a-affinity chromatography. Proteomics. 2001;1:773-777.
- Alton G, Cox AD, Toussaint LG 3rd, Westwick JK. Functional proteomics analysis of GTPase signaling networks. Methods Enzymol. 2001;332:300-316.
- Hooper NM. Determination of glycosyl-phosphatidylinositol membrane protein anchorage. Proteomics. 2001;1:748-755.
- Faulkner S, Dun MD, Hondermarck H. Proteogenomics: emergence and promise. Cell Mol Life Sci. 2015;72:953- 957.
- Rivers RC, Kinsinger C, Boja ES, Hiltke T, Mesri M. Linking cancer genome to proteome: NCI’s investment into proteogenomics. Proteomics. 2014;14:2633-2636.
- Zhang B, Wang J, Wang X, Zhu J, Liu Q. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382-387.