Research Article
Bioinformatic Evaluation and Comparison of Parallel aSNP and aCGH Analyses of Myelodysplastic Syndromes Patients with Normal Karyotype
Sabrina Claßen-von Spee1, Mar Mallo2, Manfred Beier1, Simone de Leve1, Leonor Arenillas3, Carmen Pedro4, Francesc Solé2 and Brigitte Royer-Pokora1*
1Institute of Human Genetics, Heinrich-Heine University, Duesseldorf, Germany
2MDS Research Group, Institut de Recerca Contra la Leucèmia Josep Carreras, Barcelona, Spain
3Laboratori de Citologia Hematològica Servei de Patologia, Hospital del Mar, Barcelona, Spain
4Servei d'Hematologia Clínica, Hospital del Mar, Barcelona, Spain
*Corresponding author: Brigitte Royer-Pokora, Institute of Human Genetics, Heinrich-Heine University of Duesseldorf, Postfach 101007, D40001 Duesseldorf, Germany
Published: 19 Oct, 2016
Cite this article as: Claßen-von Spee S, Mallo M, Beier
M, de Leve S, Arenillas L, Pedro C,
et al. Bioinformatic Evaluation and
Comparison of Parallel aSNP and
aCGH Analyses of Myelodysplastic
Syndromes Patients with Normal
Karyotype. Clin Oncol. 2016; 1: 1118.
Abstract
To study MDS bone marrow samples for tumor specific alterations two different microarray
platforms, aSNP and aCGH, have been widely used. The purpose of this study was 1) to compare the
two array methods and 2) evaluate the usefulness of different aCGH algorithms for the identification
of authentic alterations in tumoral samples.
Parallel aSNP and aCGH analyses were performed on the same 21 bone marrow DNA samples
from karyotypically normal MDS patients. FISH and Q-PCR methods were used to verify several
alterations. The aSNP data were evaluated using Genotyping Console Software; aCGH data were
analyzed with the ADM-2 setting of the Agilent Genomic Workbench program, followed by three
additional algorithms, haarseq, lawsglad and dnacopy. 404 alterations were seen with aSNP of these
74 were also seen with aCGH with at least the ADM-2 algorithm. With the ADM-2 setting, 237
imbalances were detected, of these 72 were seen with all four aCGH algorithms. Among the latter
aberrations were two tumour specific deletions, a TET2 deletion and a larger deletion containing
DNMT3A, present in a high percentage of cells. One tumour specific telomeric 16p gain only seen
with aCGH was confirmed with FISH in 7.5% of the cells. As expected, uniparental disomies (UPDs)
were only detected with aSNP; in one case at 3q and in the other case two UPDs at 4q and 5p. The
discrepancies between both methods and the algorithms are discussed in detail.
Our results show that 72/237 (30%) aCGH alterations were predicted with all four algorithms. Of the
74 alterations seen with both platforms 31 were seen with all algorithms. 18% of the aSNP alterations
and 31% of the aCGH alterations were also seen with the other platform. Of 15 selected aberrations
detected with aSNP only and with the highest deviation from normal 50% could be confirmed by
Q-PCR, whereas all 10 selected imbalances detected with aCGH only were confirmed by Q-PCR.
Therefore, using several algorithms for aCGH analysis, increases the number of true alterations.
aSNP data should be interpreted with caution and another verification method is advisable.
Keywords: aSNP; aCGH; Karyotypically normal MDS; Bioinformatic evaluation
Introduction
The Myelodysplastic Syndromes (MDS) are a heterogeneous group of clonal disorders in
the haematopoietic system. Characteristic for MDS is an ineffective haematopoiesis, dysplasia,
various degrees of cytopenia and a risk to evolve in Acute Myeloid Leukaemia (AML). For risk
calculation the international prognostic scoring system (IPSS) [1] is used and, recently, the IPSS-R
has been introduced [2]. In this new IPSS-R, the patients are divided into five risk groups for AML
transformation and survival (very low, low, intermediate, high and very high), depending on clinical
parameters and the karyotype, being the parameter with the strongest impact [2,3,4]. In both
systems, the IPSS and IPSS-R, a cytogenetic normal karyotype often leads to the allocation into a
better (good or int-1) risk group and 40-50% of the patients show a normal bone marrow karyotype.
Classical cytogenetic analyses are limited by their resolution and the need of mitotic cells, which
is not always successful in MDS. Therefore, array CGH and SNP microarrays were used to analyze MDS bone marrow samples.
However, not in all microarray studies exclusively cytogenetically
normal cases were included and in some cases it was stated that
cytogenetic analyses were not successful. Therefore, it is not surprising
that in some studies a simple addition of all genomic imbalances
correlates with a shorter survival as in some cases cytogenetic
abnormalities most likely were present. Nevertheless, cryptic
imbalances were useful for better prognosis prediction [5,6,7]. In
our previous aCGH study of exclusively MDS patients with a normal
karyotype we found that 42 of 107 (45%) showed hidden genomic
aberrations and many of these were verified by other methods [8].
The patients with hidden additional imbalances had a shorter survival
than patients without sub microscopic alterations [8].
The study of bone marrow samples of MDS patients is specifically
challenging as per definition only maximally 20% of the bone marrow
cells are blast cells. Therefore, some authors used CD34+ enriched
cell populations but this is a more time consuming method and only
small amounts of DNA can be obtained. We therefore compared
aCGH and aSNP for their efficiency to detect cryptic aberrations in
MDS with normal karyotype using the same DNA from bone marrow
samples. The aCGH results were evaluated using four algorithms and
the results were compared.
Materials and Methods
Patients and samples
A total of 21 patients with primary MDS and a normal
karyotype were included. MDS diagnoses were made according to
the 2008 World Health Organization (WHO) [9] classification. The
cytogenetic analyses were performed by one of us (FS) and normally
20 metaphases were analyzed. Diagnoses and IPSS scores for these
patients are shown in Additional file 1: Table S1. The study design was
approved by the Institutional review board (CEIC-Comité Ético de
Investigación Clínica Parc de Salut MAR (no. 2008/3268/I)) before its
initiation. Informed consent was obtained from all patients enrolled
in the study in accordance with the Declaration of Helsinki.
Tumour DNA was isolated from whole bone marrow. All samples
were obtained from Parc de Salut Mar Biobank, Mar Biobanc. Germline
DNA from eight patients was extracted from isolated peripheral
blood CD3+ T-cells (MACS, Miltenyi Biotec GmbH, Germany). DNA
was extracted with the Gentra Puregene kit (Qiagen Inc, Valencia,
CA, USA). The purity and concentration of genomic DNA was
evaluated using the ND-1000 Spectrophotometer (Thermo Fisher
Scientific, Wilmington, DE, USA). The integrity was checked by a 2%
agarose gel.
Array platforms and evaluations
For the aSNP analyses we used the Genome-Wide Human
SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA). Analyses were
performed as described in the protocol by the manufacturer (P/N
702607 rev. 2, Affymetrix). Only DNA that fulfilled quality controls
required by Affymetrix was submitted for array procedure. Briefly,
500 ng of genomic DNA was digested with restriction enzymes.
Then, fragments were ligated to the adapter and amplified by
polymerase chain reaction, purified by magnetic beads, fragmented
and end-labelled using terminal deoxynucleotidyl transferase.
Labelled fragmented amplicons were hybridized to the SNParrays
and after washing and staining in a Fluidics Station, arrays
were scanned, data analyzed using Genotyping Console Software
Version 4.0, Chromosome Analysis Suite Version 1.0.1 (Affymetrix),
using annotations of genome version NCBIv30 (hg18). Only those
achieving manufacturers’ quality cut-off parameters were included
in the analysis. In addition to software-reported CNAs of 100 Kb
that carried a minimum of 10 aberrant probes, a visual analysis was
performed. Paired sample analysis with T-cell-derived DNA was used
to identify germ-line lesions. For CN-LOH, we applied the following
threshold: ≥ 50 altered probe sets (SNPs) at least 2 Mb in size [10]
for paired sample analysis. Size and location based exclusion criteria
(interstitial ≥25 Mb and telomeric ≥50 probes in ≥2 Mb) was applied
for non-paired analysis [11].
For the aCGH analyses, we used 4x180K oligonucleotide
microarrays (Human Genome CGH Microarray, Agilent
Technologies, Palo Alto, CA, USA). Analyses were performed as
described in the protocol by the manufacturer (Protocol Version
6.2.1, February 2010; Agilent). Between 500-900 ng DNA was used
for labelling and hybridization. In all cases same-gender reference
DNA, pooled from 200 individuals was used as control (Kreatech).
In cases, where CD3+ T-cell DNA was available this was hybridized
separately with the same control reference DNA, to be able to detect
germ-line alterations. The DNA of the patients was labelled with
cyanine 5, the reference DNA was labelled with cyanine 3 and both
were hybridized simultaneously on the same slide. After scanning,
the data were analyzed using the Agilent Feature Extraction Software
(Version 10.7.1.1) and visualized with Agilent Genomic Workbench
(Version 7.0), algorithms ADM-2 and threshold 6.2. In addition,
three other analysis were used to detect aberrations. Alterations were
filtered for affecting at least three oligonucleotides and a minimal
absolute log2-ratio of 0.2.
Both aSNP and aCGH data were submitted and are available
at the Gene Expression Omnibus (GEO) database under accession
number GSE49004 (aSNP) and GSE50897 (aCGH).
Verification Methods
Imbalances >200kb identified with aCGH were confirmed with
FISH, smaller aberrations were verified by Q-PCR. One aberration
was analyzed by FISH. Therefore two FISH probes on 16p13.3 were
chosen from UCSC Genome Bioinformatics site, one probe located
inside the aberrations (RP11-20I23) and the other probe mapping
outside of the alteration (RP11-346B16). The DNA was isolated with
a plasmid DNA purification kit (Macherey-Nagel, Düren, Germany).
The probes were labelled with different dyes; DNA of RP11-346B16
was indirectly labelled with digoxigenin and the DNA of RP11-20I23
with biotin by nick-translation and both probes were hybridized
simultaneously. Digoxigenin labelled DNA was detected with anti-
DIG-fluorescein, biotin labelled DNA was detected with streptavidincyanine
3. At least 200 cells were evaluated and a control sample was
hybridized to determine the cut off for the respective BAC probes
[12].
For the smaller aberrations, specific Q-PCR primers were
designed (Additional file 2: Table S2). The Q-PCR was done with
the Fast Start Universal SYBR Green Master Mix from Roche
(Roche Applied Science, Mannheim, Germany). The analyses were
done in triplicates and every run was repeated at least once. From
these six values the mean and standard deviation was calculated. As
controls, pooled female and male DNAs were used to calculate the
copy number. As reference we used the single copy gene PRNP. The
2-ddCt-values from the controls and the patients were calculated and compared statistically (2-sided t-test assuming equal variance of the
triplicates).
In one case a gain was verified with a custom array, designed
to cover the aberration densely including the areas where the two
presumed breakpoints were localized. This was achieved by using the
eArray platform of Agilent (http://earray.chem.-agilent.com/earray/).
The average distance of oligonucleotides within the aberration and
the putative breakpoints was 1 Kb.
Results
To make a comparison between the different array platforms the
same DNA isolated from 21 bone marrow samples was analyzed in
Barcelona with aSNP (Affymetrix) and in Duesseldorf with aCGH
(Agilent). To compare the results we used the entire set of alterations
including all known CNVs. The focus of this work was to study how
many aberrations were detected with either method alone or with
both methods and to validate the various algorithms used for aCGH
aberration calling. The difficulty in studying tumor genomes is the
mixture of cells. In the aCGH analysis we considered a germ-line
aberration if it was present in all cells, i.e. log2-ratio -1 (heterozygous
deletion) or +0.6 (heterozygous duplication). Furthermore, a normal
control sample from the same patient is desirable for UPD calling and
to rule-out germ-line alterations accurately. Therefore in this study
eight samples from isolated T-cells were used from the same patients,
as T-cells are not regarded as being part of the malignant cells in MDS
[13].
Determination of aberrant regions and testing of various
algorithms
The aCGH arrays were first analyzed with Agilent's Genomic
Workbench software version 7.0, using its ADM-2 algorithm with
default settings for breakpoint detection. The threshold for calling
aberrations was lowered from the standard setting of 0.25 to a logratio
of 0.2, which was also our cut-off for all further analyses. To
support the predictions made with the aCGH platform we applied
three more methods to the raw feature extraction data, namely the
"haarseg" algorithm [14], "lawsglad" [15] and "dnacopy" [16]. All
three methods are available as additional packages for the R Statistical
Environment [17]. Usage of the first two R-packages for breakpoint
detection without changing default values was straightforward, but
dnacopy required some tuning: to ensure that aberrations with an
absolute log-ratio above our 0.2 cut-off would be detectable, the
parameter "undo.SD" was calculated as 0.2 / DLRS (Derivative Log
Ratio Spread according to Agilent), preventing two adjacent segments
with a difference in log-ratios of 0.2 or more from being merged in the
"undo"-step of the dnacopy algorithm.
For Affymetrix aSNP data, the aberrations reported by the
Genotyping Console Software version 4.0 (Affymetrix) were
converted to Agilent calls by mapping Agilent oligonucleotide
positions to the aberrant Affymetrix regions, subsequently treating
these aberrations as if called by the mapped oligonucleotides, with a
log-ratio corresponding to the stated copy number. With 1.8 million
probes and an average distance between markers of 700 bases, the
targets of the Affymetrix platform are spotted much more densely
than those of Agilent arrays covering the human genome with only
170.000 probes, but at the same time a larger number of aberrant
spots (>10) is recommended to warrant a call. Still, even requiring at
least 10 oligonucleotides the Affymetrix array would be able to detect
much smaller regions not detectable with the Agilent platform. By converting Affymetrix calls to Agilent calls it was possible to apply
the same lower threshold of at least three Agilent oligonucleotides,
i.e. the same average minimal length of aberrant regions to both data
sets. Therefore, in this work we considered only aberrations that are
large enough to be seen with both platforms. An R-script was written
to combine the results of all five methods, merging overlapping
predictions into contiguous regions. Figure 1 shows how a “contig”
was created from the dataset. In the following, each such contig is regarded as exactly one aberration, predicted by the union of the
methods involved, but usually consisting of a number of segments
called by different combinations of these methods. Contigs with less
than three oligonucleotides were removed.
330 alterations were only detected with aSNP and 163 only with
aCGH. In addition, 74 aberrations were found with both platforms,
resulting in a total of 237 aCGH and 404 aSNP aberrations (Figure
2a). The inner circle in the Venn diagram depicts the number of
aCGH aberrations seen with all four algorithms. Of the 74 alterations
seen with both platforms 31 were seen with all four aCGH algorithms;
in addition 41 of the 163 aCGH only alterations were detected with all
four different methods, resulting in total of 72 imbalances. In summary,
82% of the alterations detected with the Affymetrix program were not
seen with aCGH (330/404), and 68% of the alterations detected with
aCGH were not seen with aSNP (163/237) (Figure 2a). Vice versa,
18% of the aSNP alterations were in common with aCGH and 32% of
the aCGH imbalances were also seen with aSNP, indicating that both
platforms have a relatively high discordance.
Figure 2b shows a more detailed analysis of the aCGH data using
the three additional programs. It can be seen that 37 aberrations were
called with ADM-2 only. Further 36 alterations were detected with
ADM-2 plus one and 92 aberrant regions were found with ADM-2
and two further algorithms (Figure 2b). Of the 72 alterations called
with ADM-2 and all three additional programs 31 were also seen with
aSNP. These are the most reliable alterations as they were called with
all four programs and both platforms.
Table 1 shows the core region of these 31 aberrations and 27 of
these correspond 100% to known CNVs. Of note one of these, the
TET2 deletion is also listed as 100% CNV in the Table, although
it is a tumor specific deletion. CNVs were identified by using the
database of genomic variants (http://dgv.tcag.ca; NCBI36_hg18_
variants_2014-10-16.txt). The imbalances that are seen with both
platforms and all four aCGH algorithms need not be verified as these
are the most reliable, although in one case a custom array verified
the small gain. The core regions of the 41 alterations detected with all
four aCGH algorithms but not with aSNP are listed in Table 2. As all Q-PCRs performed verified the imbalances we suggest that in these
cases another verification is not necessary.
To further explore the basis for the inconsistencies between
the platforms, we performed Q-PCR analyses. For this purpose we
selected aberrations either seen with all four programs in aCGH
and but not with aSNP, or apparently homozygous aberrations called as four or zero copies with aSNP but not with aCGH. A total
of 11 aberrations were thus analyzed in 15 aSNP only altered cases
and in 10 aCGH only altered cases. The positions in the genome
and the primer sequences for Q-PCR are listed in additional file 2,
Table S2 and all correspond to known CNVs. In some cases where
enough DNA from bone marrow cells was unavailable we used DNA from T-cells as CNVs should be seen in bone marrow and T-cells.
(Figure 3) shows the results for these analyses: the top panel shows
the relative intensity of aCGH, where 1.5 corresponds to three copies
(heterozygous gain), 0.5 to a heterozygous and 0.0 to a homozygous
deletion in all cells; the middle panel shows the 2-ddCt values for the
Q-PCRs relative to a probe that is present in 2 copies in the genome
(2-ddCt=1), the bottom panel shows the copy number called by aSNP.
The left side of the panel represents alterations called by aSNP only,
14 were gain-4 and one a homozygous loss. The analyzed alterations
are listed in additional file 3: Table S3. In seven cases with gain the
Q-PCR revealed an increase in the copy number. However, due to
the large confidence interval the exact copy number increase cannot
be determined. These correspond to known CNVs (e.g. 4q13.2, UGT;
3q26.1 no gene; 16q22 DPR) and at these positions the aCGH arrays
have only few oligonucleotides. Therefore, it is not surprising that
four breakpoint detection algorithms performed poorly at these
positions. In another seven cases with aSNP gain, Q-PCR could not
confirm any gain. It is interesting, that the same alteration in 4q13.2
with a gain in case MD144 was not confirmed, whereas in five other cases a gain was confirmed. The gain of 2p11.2 seen by aSNP in cases
MD160, MD69, MD121, MD140T was not verified by Q-PCR. It
was not observed in aCGH, although the segment was covered by 19
oligonucleotides supporting the notion that it is a false positive result.
In addition, the homozygous deletion in MD93H was not confirmed
with Q-PCR although the 2-ddCt level was less than 1 (Figure 3).
The right side of the panel shows 10 alterations only seen with
aCGH and all were verified with Q-PCR. For example, in case MD140,
bone marrow showed a homozygous loss of 11q11 and the loss was
verified by Q-PCR. Aberrations affecting a known CNV on 8p11.23
were seen by aCGH in cases PD160 and MD144 as a homozygous
loss and in case NM21384 as a gain, verified by Q-PCR. Figure 4a
shows the homozygous deletion in PD160 as seen in the Genomic
Workbench and Figure 4b the heterozygous gain in NM21384. Both
alterations could be verified by Q-PCR as shown in Figure 4c. In
contrast, a homozygous loss in the same region in 8p11.23 seen in
case MD93H only by aSNP could not be verified by Q-PCR, although
a slight reduction can be observed (Figure 4c and d). In case MD69,
a gain to three copies in 12p13.31 was observed by aCGH and was observed as a four copy gain by Q-PCR (Figure 3a and b). It is
interesting that none of these 10 verified imbalances were called by
aSNP, although the smallest of these regions was covered by more
than 20 oligonucleotides.
In summary the tested homozygous losses, a heterozygous loss
and two gains observed only by aCGH were verified by Q-PCR, and
gains as observed by aSNP were verified in 50% of the cases. In some
cases (e.g. 4q13.2, 16q22) these were not seen with aCGH because
these regions were not covered by enough oligonucleotides.
Larger tumor specific alterations not present in all cells
In two cases, known MDS tumor specific aberrations were found
with both platforms and all aCGH algorithms (Table 1). In case
MD44, a heterozygous deletion of 424 kb including the TET2 gene
was identified as the sole imbalance with a log-ratio of -0.85, present
in 89% of the cells. This deletion was identified with all four aCGH
evaluation programs and with aSNP and it is listed in the database of
genomic variants as CNV. Sequencing revealed a mutation in exon 5
(p.Q1191X) without a wild type allele, confirming the heterozygous
deletion. Loss of the normal allele supports its function as a tumour
suppressor gene.
In case MD72, a 901 kb deletion in 2p23.3 containing CENPO
(disrupted by the deletion), ADCY3, DNAJC27, EFR3B, DNMT3A
and DTNBL (disrupted) was revealed with all programs and both
methods. It had a log ratio of -0.76, corresponding to a heterozygous
deletion in 82% of the cells. We have observed a similar but larger
deletion in our previous series of karyotypically normal MDS patients
[8]. DNMT3A gene, often mutated in MDS/AML, resides in this
deletion. As this deletion spans a large segment, was present in a high
percentage of cells and is a known MDS associated gene, verification
was not necessary.
In one case (MD69) a tumour specific 3 Mb gain of 16p13.3
with a log-ratio of 0.19 was detected only with aCGH by two
programs (ADM-2, lawsglad) (Figure 5). The log-ratio of 0.19 was
just below our cut-off used; however, the large size with 248 affected
oligonucleotides suggests that this gain is tumor specific and present
in only a few cells. This gain was confirmed with FISH in 7.5% of the
cells (data not shown).
Furthermore we re-evaluated our previously published aCGH
cases using the three additional algorithms. For this purpose, we
analyzed tumor specific alterations that were confirmed with either
FISH or Q-PCR [8]. The two 7q22, two 5q31, two RUNX1 and two
TET2 deletions were seen with all four algorithms. However, it should
be noted that these were present in 45-95% of the cells. Therefore,
it is currently not resolved whether aberrations present in a lower
percentage of cells are always detectable with all four analysis
methods.
One small germ-line alteration identified by aCGH and not
entirely described as CNV
One gain of 695 kb in 18q22.3 with a log-ratio of 0.54 was detected
with all four aCGH programs and aSNP in case MD51 and was verified by a custom array. This alteration is present in bone marrow
and T-cells confirming that it is a germ-line gain. Several small CNVs
map to this segment; however, the entire region was not described as
variant (64%, Table 1). This segment contains several genes FBOX5
(disrupted), CYB5A, FAM69C, CNDP2, CNDP1, ZNF107 (disrupted),
that might be risk factors for MDS development. For example the
disrupted FBOXO15 gene codes for a protein with the 40-amino
acid F-box motif and may act as protein-ubiquitin ligase; an intact
gene with increased copy number, CYB5a, encodes cytochrome b5
reductase, reducing met haemoglobin (ferric haemoglobin) to normal
haemoglobin (ferrous Hb). One patient with the autosomal recessive
disease type IV hereditary methaemoglobinemia was described with
a homozygous splice mutation in this gene [18]. Another gene with
increased copy number, CNDP2 encodes tissue carnosinase/with
a putative function in glutathione metabolism and in xenobiotic
metabolic processes. This gene could play a role after exposure to toxic
substances, known to be risk factors for MDS development. The same
alteration was not observed in any of 515 cases that we have analyzed
with the same array type (unpublished observation). Therefore it is
possible that the germ-line gain affecting one or several of the genes
in this novel 695 kb gain could contribute to MDS development.
UPDs seen with SNP arrays
The detection of UPDs can be helpful to identify new putative
genes involved in MDS or other malignancies [19]. UPDs containing
relevant genes implicated in cancer were detected in two cases (2/22,
9%) (Figure 6). In case MD116 one UPD was found in 3q and in case
MD117, two UPD regions were identified in 4q and 5p (Figure 6).
It is interesting that these two cases did not have any other tumour
specific gains or deletions. Several genes related to tumour growth
map to these segments, for example the 3q25qter region spans
38 Mb and contains several important genes involved in tumour
development such as PIK3CA, EVI1/MDS, BCL6 and ETV5 and in
apoptosis for example TNFS10. Several tumor related genes map to
the UPD on 4q, e.g. CDKN2AIP, a novel regulator of the p53 pathway,
ING2 (inhibitor of growth), modulating histone acetyltransferase and
histone deacetylase complexes and with a function in DNA repair
and apoptosis, CASP3, encoding a protein involved in the apoptotic
cell both by extrinsic (death ligand) and intrinsic (mitochondrial)
pathways, MLF1, a factor required for centromere assembly and the
tumour suppressor gene FAT1. The UPD region in 5p15 contains
TERT, encoding a catalytic subunit of the enzyme telomerase with
an important function in maintaining the chromosome ends, NKD2
a negative regulator of Wnt receptor signalling and two homeobox
genes, one with a possible tumour suppressor function in gastric
cancer (IRX1).
Figure 1
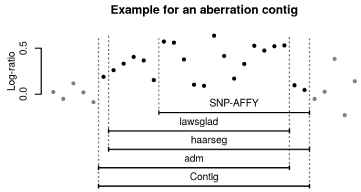
Figure 1
Creation of a “contig” from the database. The overlapping regions
of all programs used for aCGH and aSNP data analysis were merged into
contigs. The contigs called in the analyzed region by the four programs were
treated as one cohesive aberration/contig.
Figure 2
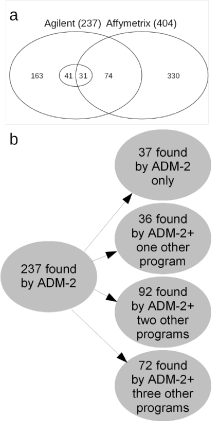
Figure 2
Overview of the numbers of aberrations found with aSNP and/or
aCGH. (a) Venn Diagram of the aberrations found by the array analyses.
With aSNP a total of 404 alterations were found, 74 of these were also seen
by aCGH and 31 of these with all four algorithms (centre circle of diagram).
A total of 237 aberrations were seen by aCGH analyses using the Agilent
Genomic Workbench 7.0, ADM-2 algorithm. 72 were seen with ADM-2 and
all of the three statistical programs; 31 of these were also seen with aSNP.
(b) Details of aCGH aberrations identified by the different algorithms. Of 237
aberrations 37 were seen only with ADM-2, 36 with AMD-2 and one further
program, where as 92 were detected by ADM-2 and two other programs.
Lastly, 72 alterations were seen by ADM-2 and all three statistical programs.
Table 1
Table 1
Core regions of the aberrations found by aCGH and aSNP.
Table 2
Table 2
Core regions of the aberrations found with aCGH only.
Figure 3
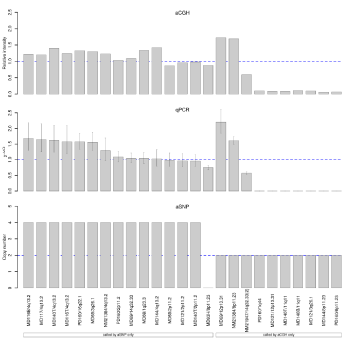
Figure 3
Comparison of alterations detected only with either method and Q-PCR results. The left side of the panels shows aberrations found only by aSNP and the
right side, those only seen with aCGH (all four algorithms). (a) Chosen aCGH aberrations with their relative intensity. A value of 1.5 indicates three copies, a relative
intensity of 0.0 shows a homozygous deletion. (b) Q-PCR results shown as 2-ddCt-values, a value of 1.0 indicates two copies, whereas 0.5 indicates a heterozygous deletion, and 1.5 a gain of one copy. 0.0 shows a homozygous deletion, i.e. both copies deleted and 2.0 a gain of two copies, i.e. four copies in total. (c) Results of aSNP analyses (copy number) are shown. Loss of both copies leads to a copy number of 0, gain of two copies results in a copy number of 4. Copy number of
two indicates that two copies are present (normal situation).
Figure 4
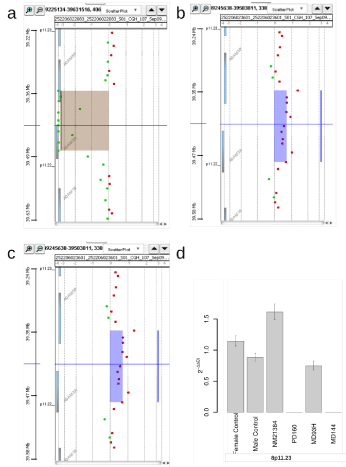
Figure 4
Different aberrations of 8p11.23, found by aSNP or aCGH, shown in Geneview and the Q-PCR results. (a) In PD160 aCGH detected a loss of both copies (homozygous) of 8p11.23 as shown in Geneview, aSNP did not detect any loss in this region. (b) aCGH analysis of NM21384 showed a gain in 8p11.23, resulting in three copies as shown in Geneview. (c) Q-PCR results with primers located within the ADAM3A gene. The heterozygous gain of 8p11.23 in case NM21384 (2-ddCt: 1,61) and the loss of both copies in this region in case PD160 (2-ddCt: 0) could be verified by Q-PCR. In case MD93, a homozygous loss as found by the aSNP
analysis could not be verified, instead Q-PCR resulted in a 2-ddCt-value of 0.74. The 95% confidence interval is indicated. (d) aSNP showed a loss of both copies of 8p11.23 in case MD93, but aCGH did not detect this aberration as seen here with Agilent Workbench 7.0, ADM-2.
Figure 5
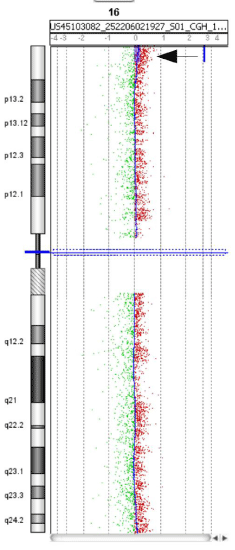
Figure 5
Overview of chromosome 16 of case MD69. A 3 Mb gain of 16p13.3
was detected as shown here in chromosome view, Genomic Workbench 7.0,
Agilent. This was called by lawsglad and ADM-2 (position: 46270-3065196).
The gain showed a log-ratio of +0.19 and could be verified by FISH in 7.5%
of the cells.
Discussion
In this study of MDS with normal karyotype, we compared two
different array platforms (Agilent vs. Affymetrix) using the same
DNA samples in two different labs. In addition to the standard
Agilent Workbench program, used in most laboratories for the
evaluation of aCGH data, three further algorithms were applied
and their usefulness was determined. With the default setting of the
Agilent Workbench program ADM-2 and a lower log-ratio cut off of
0.2, 237 aberrations were detected. Adding more programs for the
evaluation of the data reduced the number significantly and only 72
were called with all four algorithms used. It is not surprising that most
of these correspond to CNVs present in all cells. However, two tumor
specific deletions present in a high percentage of cells were among
these and one was labelled 100% and the other as 80% CNV by the
DGV database. Clustering of the aCGH methods showed that ADM-
2 and lawsglad had the highest concordance (Figure 7). Therefore, it is
advisable adding at least lawsglad to the standard Agilent program to
improve the reliability of the analysis for identifying true aberrations.
It is much more challenging to identify alterations present only in
a low percentage of cells as would be expected in a mix of cells in MDS
bone marrow. Only three tumor specific imbalances were detected
and it is interesting that one was found in 86% and the other in 93%,
while the third was only present in 7.5% of the total bone marrow
cells. In our previous series of karyotypically normal MDS cases we
also found tumor specific aberrations in a high percentage of cells [8].
The current reanalysis of eight tumor specific alterations from this
previous work [8] revealed that all were seen with the three additional
algorithms. The terminal gain in 16p in 7.5% of the cells and with a
log-ratio of 0.19 was seen with ADM-2 and lawsglad, but not with
the other two programs. This shows that tumor specific imbalances when present in a low percentage of cells, may not be called by all
algorithms but could nevertheless be true imbalances. However, we
have analyzed two small aberrations called with ADM-2 and one
additional algorithm that could not be verified by Q-PCR. In both
cases the log-ratio of 0.4 suggested that about 50% of the cells have a
heterozygous deletion and this should have been detectable. Of note,
if an alteration is present in a very low percentage of cells it is difficult
to confirm with Q-PCR as the sensitivity is not high enough. In such
cases we have successfully used custom arrays, covering the region
of interest densely with oligonucleotides to verify the aberration [8].
Thus an aberration present in a low percentage of cells can be verified
with another method.
It is interesting to point out, that in our previous studies of MDS,
we did find that all hidden additional imbalances were present in a
high percentage of bone marrow cells either by FISH (between 17 and
96%) or Q-PCR [8]. In addition, we also showed that the 5q deletions
were present in 42-91% of total bone marrow cells, although the
number of bone marrow blasts ranges between 1 and 30% [20]. This is
due to the presence of mature cells in the total bone marrow that also
harbour the genetic abnormalities. These studies showed that tumor
specific imbalances in MDS are usually found in a higher percentage
of cells.
The aSNP data were evaluated with the Affymetrix’ software and
of the 404 aberrations (without UPDs) only 74 aberrations were also
seen with aCGH (18%), demonstrating that a large percentage of the
detected abnormalities were not observed with the other platform.
Of the 15 selected aSNP alterations with the highest level of gain or
loss not seen with aCGH only half could be confirmed by Q-PCR.
The higher number of additional imbalances observed with aSNP
only, can partially be explained by the fact that aCGH arrays have few
oligonucleotides in many known CNV regions and these are therefore
not detected. This was observed in the Q-PCR analyses as several of
the confirmed imbalances resided in those CNV regions not covered
by aCGH oligonucleotides. This would also suggest that a number of
aSNP only imbalances not detected by aCGH map to known CNVs.
In contrast, seven tested homozygous losses, one heterozygous loss
and two heterozygous gains only seen with aCGH were confirmed. It
is therefore sensible to verify somatic imbalances detected with aSNP.
Another important, so far not completely resolved issue, is whether
these hidden alterations have the same prognostic significance as
karyotypically detected abnormalities [21]. In light of our results
it is important to remember that in many array studies imbalances
detected by aSNP were not verified by other methods. Furthermore,
as cases were included with unsuccessful cytogenetics, the "hidden" imbalances might have been detected if karyotype analysis would
have been successful. This could explain why often high frequencies
of "hidden" aberrations are described in these studies, whereas in this
report we have used only karyotypically normal cases. We found two
tumor specific deletions and one gain and two cases with UPD, i.e.
5/21 (23.8%) cases had additional hidden imbalances. Therefore, in
order to determine the correct prognostic significance of these hidden
aberrations large series of cases with a successful normal karyotype
should be studied in the future with verification of at least some of the
imbalances. In addition, another question that needs to be addressed
is the identity of the gene/s affected by the aberration. For example
if either TET2 or TP53 are deleted this will have a different impact
for prognosis, while some reports merely count the size of the total
genomic imbalances [22,23]. Furthermore, another question is
whether a single hidden aberration might have a different impact
than several hidden imbalances as seen here in the case with two UPD
segments. On top of all these questions we have shown here that not
all alterations detected with different platforms or algorithms can be
confirmed with other methods.
Figure 6
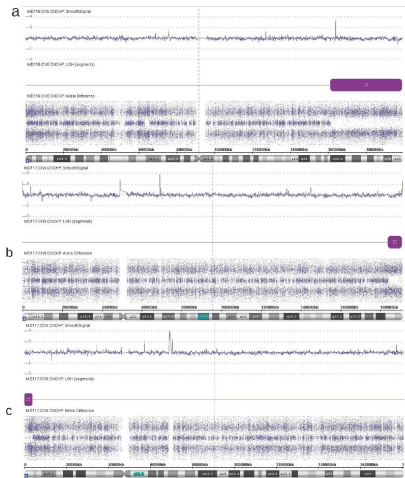
Figure 6
Three UPD regions identified in two cases with aSNP. (a) Case MD116 with UPD of chromosome 3q. (b) UPD of 4q in case MD117. (c) UPD of
chromosome 5p also in case MD117.
Figure 7
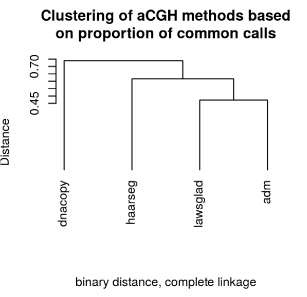
Figure 7
Clustering of aCGH methods based on proportion of common calls.
The hierarchical cluster analysis showed that the results of lawsglad and
ADM-2 are highly concordant, whereas dnacopy and ADM-2 had the lowest
concordance.
Conclusion
In summary, we show here that of 567 combined imbalances from two different platforms with the same DNA samples 74 (13%) are in common. The aCGH studies revealed less aberrations than aSNP, suggesting that the latter platform could result in more false positives or could detect more aberrations. The data from the aSNP platform with a higher number of alterations should be interpreted with caution and verification with other methods are also advisable, when these are not present in all cells. Maybe studies with different aSNP algorithms are required. Using different analysis aCGH algorithms improves the reliability for the aCGH results and reduces the number of highly likely true alterations. Therefore one or two other analysis methods should be included in addition to ADM-2. Verification of alterations found with four aCGH analysis methods is not necessary. In contrast, for aCGH alterations found in a lower percentage of cells and with less that three methods another verification is advisable.
Acknowledgement
We thank Dr. Barbara Hildebrandt, Institute of Human Genetics, Duesseldorf for help with the FISH analysis. This collaborative work was initiated through the activities of COST Action BM0801: European Genetic and Epigenetic Study on AML and MDS.
References
- Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997; 89: 2079-2088.
- Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012; 120: 2454-2465.
- Schanz J, Steidl C, Fonatsch C, Pfeilstöcker M, Nösslinger T, Tuechler H, et al. Coalesced multicentric analysis of 2,351 patients with myelodysplastic syndromes indicates an underestimation of poor-risk cytogenetics of myelodysplastic syndromes in the international prognostic scoring system. J Clin Oncol. 2011; 29: 1963-1970.
- Schanz J, Tüchler H, Solé F, Mallo M, Luño E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. Clin Oncol. 2012; 30: 820-829.
- Mohamedali A, Gäken J, Twine NA, Ingram W, Westwood N, Lea NC, et al. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood. 2007; 110: 3365-3373.
- Heinrichs S, Kulkarni RV, Bueso-Ramos CE, Levine RL, Loh ML, Li C, et al. Accurate detection of uniparental disomy and microdeletions by SNP array analysis in myelodysplastic syndromes with normal cytogenetics. Leukemia. 2009; 23: 1605-1613.
- Tiu RV, Gondek LP, O'Keefe CL, Elson P, Huh J, Mohamedali A, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011; 117: 4552-4560.
- Thiel A, Beier M, Ingenhag D, Servan K, Hein M, Moeller V, et al. Comprehensive array CGH of normal karyotype myelodysplastic syndromes reveals hidden recurrent and individual genomic copy number alterations with prognostic relevance. Leukemia. 2011; 25: 387-399.
- Brunning RD, Orazi A, Germing U, Le Beau MM. Myelodysplastic syndromes/neoplasms, overview. In: Swerdlow SH, Campo E, Lee Harris N, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO classification of Tumours of Haematopoietic and Lympoid Tissues. 2008 Lyon: IARC Press. 88-107.
- Heinrichs S, Look AT. Identification of structural aberrations in cancer by SNP array analysis. Genome Biol. 2007; 87: 219.
- Maciejewski JP, Tiu RV, O'Keefe C. Application of array-based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br J Haematol. 2009; 146: 479-488.
- Trost D, Hildebrandt B, Müller N, Germing U, Royer-Pokora B. Hidden chromosomal aberrations are rare in primary myelodysplastic syndromes with evolution to acute myeloid leukaemia and normal cytogenetics. Leuk Res. 2004; 28: 171-177.
- Miura I, Takahashi N, Kobayashi Y, Saito K, Miura AB. Molecular Cytogenetics of Stem Cells: Target cells of Chromosome Aberrations as Revealed by the Application of Fluorescence In Situ Hybridization to Fluorescence-Activated Cell Sorting. Int J Hematol. 2000; 72: 310-317.
- Erez Ben-Yaacov and Yonina C. Eldar. HaarSeg: HaarSeg. R package version 0.0.3/r4. 2009.
- Philippe Hupe. GLAD: Gain and Loss Analysis of DNA. R package version 2.34.0. 2011.
- Venkatraman E. Seshan and Adam Olshen. DNAcopy: DNA copy number data analysis. R package version 1.44.0.
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2015.
- Giordano SJ, Kaftory A, Steggles AW. A splicing mutation in the cytochrome b5 gene from a patient with congenital methemoglobinemia and pseudohermaphrodism. Hum Genet. 1994; 93: 568-570.
- Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res. 2011; 17: 3913-3923.
- Evers C, Beier M, Poelitz A, Hildebrandt B, Servan S, Drechsler M, et al. Molecular definition of chromosome arm 5q deletion end points and detection of hidden aberrations in patients with myelodysplastic syndromes and isolated del (5q) using oligonucleotide array CGH. Genes Chromosomes Cancer. 2007; 46: 1119-1128.
- Arenillas L, Mallo M, Ramos F, Guinta K, Barragán E, Lumbreras E, et al. Single nucleotide polymorphism array karyotyping: a diagnostic and prognostic tool in myelodysplastic syndromes with unsuccessful conventional cytogenetic testing. Genes Chromosomes Cancer. 2013; 52: 1167-1177.
- Cluzeau T, Moreilhon C, Mounier N, Karsenti JM, Gastaud L, Garnier G, et al. Total genomic alteration as measured by SNP-array-based molecular karyotyping is predictive of overall survival in a cohort of MDS or AML patients treated with azacitidine. Blood Cancer J. 2013; 3: 155.
- Starczynowski DT, Vercauteren S, Telenius A, Sung S, Tohyama K, Brooks-Wilson A, et al. High-resolution whole genome tiling path array CGH analysis of CD34+ cells from patients with low-risk myelodysplastic syndromes reveals cryptic copy number alterations and predicts overall and leukemia-free survival. Blood. 2008; 112: 3412-3424.