Research Article
Disturbed B cell and DC-Homeostasis in Pediatric cGVHD Patients-Cocultivation Experiments and Review of the Literature
Julian Zipfel*, Matthias Eyrich, Paul-Gerhardt Schlegel and Verena Wiegering
Department of Pediatric Hematology/Oncology and Stem Cell Transplantation, University Hospital Würzburg, Germany
*Corresponding author: Julian Zipfel, Department of Pediatric Hematology/Oncology and Stem Cell Transplantation, University Hospital Würzburg, Josef-Schneider-Str. 2, 97080 Würzburg-Germany
Published: 16 Sep, 2016
Cite this article as: Zipfel J, Eyrich M, Schlegel P-G,
Wiegering V. Disturbed B cell and
DC-Homeostasis in Pediatric cGVHD
Patients-Cocultivation Experiments and
Review of the Literature. Clin Oncol.
2016; 1: 1097.
Abstract
B cells and DCs are suspected to play an important role in the pathogenesis of cGvHD, which is a serious complication of HSCT with high morbidity. It is characterized by immune responses of donor immune cells against recipient-derived antigens. Pathogenesis is not yet fully understood, however reconstitution of B cells after HSCT has similarities to physiologic ontogeny. Immunophenotyping and co-cultivation-experiments of B cells and DCs from pediatric patients with cGvHD as well as healthy donors were conducted. Significant differences between patients and healthy donors were observed with increased memory, transitional, CD69+ and CD86+ phenotype and lower levels of naïve B cells due to apoptosis. Co-cultivation revealed this to be primarily B cell-dependent without major effects of and with DCs. There was a decreased CD11c- phenotype in patients and less apoptosis of DCs. Our data suggest a disturbed homeostasis in B cells with increased memory phenotype in patients, whereas DCs could not influence these differences, therefore DCs are not imposing as promising targets. B cell-dependent approaches should be further investigated.
Introduction
Allogeneic HSCT is an established therapy for high-risk malignancies such as relapsed leukemia
and lymphomas, but also for various severe non-malignant diseases. GvHD is a complication of
hematopoietic stem cell transplantation, with donor immune cells forming responses against host
antigens, resulting in an immunological rejection of the recipient's tissues. The reaction of donorderived
immune cells against the recipient's malignant cells describes the graft-versus-tumor or
-leukaemia (GvL) effect [1] and composes the curative potential of HSCT. Responses against host
antigens and immunological rejection of the recipient’s tissues describe Graft-versus-host disease
(GvHD). Two distinct forms of GvHD can be differentiated. Initially, acute GvHD had been defined
as occurring within the first 100 days, chronic GvHD (cGvHD) after 100 days post-HSCT. With a
deeper knowledge of the pathogenesis of both acute and chronic GvHD though, and with a better
understanding of the differences on cellular and clinical levels, the definition has been extended, as
pathogenesis seems to be different. Incidence of cGvHD may continuously follow aGvHD, occur
after resolved aGvHD or develop de novo. Various complications can arise from the disease and its
management still constitutes a big challenge.
Pediatric patients with cGvHD show significantly lower physical functioning than HSCT
recipients without cGvHD [2], impairment of growth and loss of quality of life. Even if cGvHD
is known to increase GVL effects, this benefit is outweighed by the increased mortality associated
with the disease [3,4]. Consequently, the primary goal of post-transplantation therapy is to prevent
GvHD while enhancing desired GvL effects, thus increasing survival and decreasing disease-relapse.
Still today, in spite of more than 50 years of experience with HSCT and about 25,000 allogeneic
HSCTs each year, [5] cGvHD is, after disease-relapse, the most important reason for post-transplant
mortality [6]. Up to 70% of adult patients develop cGvHD [1], while the incidence in children is lower
(20-50%), whereas rates have increased since the use of peripheral blood stem cells [7] and donors
unrelated to the patients [6]. Known risk factors for the development of cGvHD include previous
aGvHD, stem cell source (PBSC), increasing age of host, CMV-status, TBI and sex-mismatch [6].
However, the concrete mechanisms of the development of cGvHD are still not entirely known.
While pathogenesis of aGvHD has been studied extensively and seems to be predominantly T cellmediated,
in recent studies the significance of B cells in cGvHD pathogenesis has been in focus
[8]. Hypotheses exist, that cGvHD is a mainly Th2-mediated disease with autoimmune features
[9]. The possible essential steps of its pathogenesis could be triggered
by damage to the thymus following conditioning prior to HSCT or,
more significantly, aGvHD. Thus, a decrease in negative selection
of CD4+ T cells may occur, leading to a deviated immune response
with increased production of IL-4, IL-5 and IL-11, being part of
the Th2 cytokine pattern. Tissue fibroblasts can be stimulated both
directly by Th2-released IL-2, IL-10 and TGFβ1, and indirectly via
activation of macrophages, which produce PDGF and TGFβ1. This
process can be enhanced by low Treg levels resulting in further
deviation of the immune system towards Th2 and Th17 response
[10]. Th17 cells, a CD4+ T cell subset expressing the IL-23-receptor
[11], can act as B cell helpers and have been shown to increase the
formation of germinal centres as well as promoting proliferation and
activation of B cells with their signature cytokines IL-17, IL-21 and
IL-22 [12]. This dysregulation of B cell homeostasis can, as well as
high levels of BAFF, result in B cell autoreactivity [10]. In summation,
all these steps contribute to systemic effects similar to autoimmune
diseases affecting multiple organs as skin, nails, mouth, eyes, muscles,
gastrointestinal tract, liver, lungs, kidneys, heart and bone marrow
[13-16]. However, an improvement of therapy opportunities is
desperately needed, as standard therapy consisting of glucocorticoids
with or without tacrolimus or ciclosporin [1], has many side effectsespecially
in pediatric patients [6] and we are still lacking specific
therapies. Several biomarkers have been introduced and proposed
for the identification of patients at risk of cGvHD, including Tregs, B
cells, BAFF, IL-10 [17], as well as development of HY antibodies [18].
While various ways of interaction between DCs and T cells not
only via antigen presentation are known [19], their influence on B
cells has just lately become the focus of investigation. Indeed, many of
the aforementioned mechanisms directly involved in the pathogenesis
of autoimmunity are complemented or redundantly synergize
with effects of DCs and B cells on each other. In response to TLR
stimulation or BCR ligation, pDCs can enhance auto reactive B cell
proliferation and production of anti-snRNP auto antibodies as well as
B cell survival [20]. Furthermore, dendritic cells have been shown to
be the most important APCs. In mouse models anti-CD40 activated
B cells were unable to fully prime CD4+ T cells in vivo without DCs
expressing the correct restriction element MHC II I-E [21]. This would
rather implicate an auxiliary role of B cells in optimizing CD4+ T cell
priming. Therefore only concerted presentation of antigens by both
dendritic and B cells leads to an optimal and effective CD4+ T cellimmunity
in-vivo [21]. Additionally, pDCs can induce differentiation
of CD40-activated B cells into antibody-producing plasma cells via
sequential secretion of IL-6 and type I IFN [22,23]. This explains, how
these cytokines, together with BAFF may partially contribute to the
effects of pDCs on B cell activation in addition to cell-to-cell contact
[20,24]. Besides these means of communication, a direct involvement
of the ICAM-1-LFA-1 pathway in the signaling between pDCs and
auto reactive B cells has been described [20]. As for type I IFN, it is able
to upregulated CD38 on B cells and enhance secretion of BAFF and
APRIL by B cells, monocytes and mDCs, confirming its importance
in the context of cGvHD [25,26]. Follicular DCs are involved in the
development of somatically mutated and switched memory cells [27].
Following activation via TLR7/8-L and, less pronounced, via TLR9,
pDCs have been shown to be able to induce upregulation of BLIMP1
and X-box binding protein 1, to increase B cellproliferation as well
as the differentiation into CD27high plasmablasts [28] which indicates
their involvement in B cellhomeostasis.
The aims of our study were to compare patterns of B cells and
DCs from patients with cGvHD and healthy donors. Furthermore,
apoptosis was measured and co-cultivation was used to observe
possible interactions and evaluate the effects of B cell activation on
immune cells.
Table 1
Table 1
Materials and Methods
We performed immunophenotyping of B cells and DCs from
7 pediatric patients (Table 1) with cGvHD and 13 healthy donors.
Mononuclear cells were isolated from peripheral blood via Ficoll,
followed by positive selection of CD19+ and CD14+ cells via MACS
(Miltenyi Biotec, Bergisch Gladbach, Germany). B cells were
cultivated with IL-10 (Sigma, Taufkirchen, Germany), CD14+ cells
matured and differentiated to fast DCs within 48 hours with GMCSF,
IL-4, IL-1b, IL-6, TNFα (CellGenix, Freiburg, Germany) and
PGE2 (Pharmacia Limited, Kent, UK) [29].
Cocultivation
Co-cultivation of B cells and DCs was performed in order to
determine effects of cell-type and -origin. At day 3 co-cultivation of
B cells and DCs was initiated based on characterization of the cells,
namely auto-auto (B cells patient + DCs patient), auto-allo (B cells
patient + DCs donor), allo-auto (B cells donor + DCs patient) and
allo-allo (B cells donor + DCs donor).
B cell activation
Additionally, we investigated B cell activation by further
dividing our samples into 4 equal parts and adding anti-IgM
(Jackson Laboratories, Pennsylvania, USA), CpG and Gardiquimod
(InvivoGen, San Diego, USA) or leaving blank at days 3 and 4 (Figure
1). Immunophenotypingwas performed at days 1, 3, 4 and 5.
Statistical analysis
All p-values were two-sided, considered significant below .05
and determined via independent T-tests. We compared the means
of relative proportions of cells determined via immunophenotyping.
All calculations were performed with SPSS (version 20.0.0; IBM,
Ehningen).
Figure 1
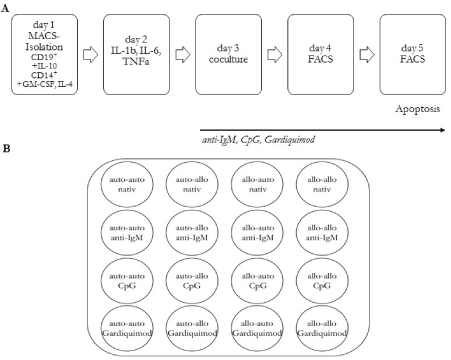
Figure 1
Experiment setting (A) day 1: Isolation of CD19+ and CD14+ mononuclear cells via MACS, adding respective agents for B cell culture und DCmaturation,
FACS day 2: adding agents for DC-differentiation, day 3: cocultivation, adding B cell activation agents and FACS with Apoptosis measurement, day 4:
repeating B cell activation and FACS, day 5: FACS with Apoptosis measurement and Co-cultivation schematic (B) with 16 well design for co-cultivation.
Results
B cells
No considerable differences between patients and healthy
donors were observed at days 1 and 3. Not until 96 hours after cell
culture were we able to measure significantly diverse cell counts.
An increased transitional, CD69+ and CD86+ (Figure 2) as well as
decreased mature naïve phenotype was observed at days 4 and 5 in
patients with cGvHD compared to healthy controls. Higher levels of
CD34+ memory cells and lower levels of BAFF-R+ naïve cells were
measured at day 5, coincident with increased apoptosis of naïve and
non-switched memory B cells (Table 2).
When taking the co-cultivation into consideration we can see
these observations confirmed. In none of the subsets do we see a
significant difference in cell counts between patients and auto-allo,
indicating no effect of DC-origin on B cells. Similarly, we did observe
significant differences between healthy donors and auto-allo in every
B cell subset except for BAFF-R+naïve B cells (Table 3).
DCs
We observed a decreased CD11c- phenotype in patients at
day 5 and less apoptosis in DCs. Other DC subsets did not differ
significantly (Table 2).
Co-cultivation reveals significant differences only between
healthy donors and auto-allo (Table 3).
Figure 2
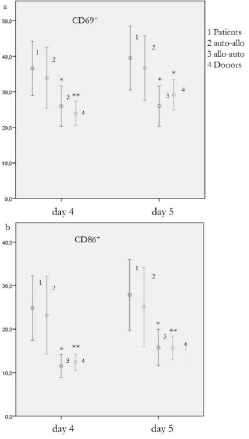
Figure 2
Comparison of percentages of CD69+ and CD86+ B cells in cocultures
with 95% confidence interval: a CD69+ B cells b CD86+ B cells, *
p-value < .05, ** p-value ≤ .001
Table 2
Table 2
Discussion
Interestingly, reconstitution of B cells after HSCT has similarities
to physiologic ontogeny [30] yet, the B cell compartment in patients
after HSCT differs from that of healthy adults [31]. Compared to
other immune cells, reconstitution of B cells is the slowest, with
reduced levels in up to 31% of pediatric patients even 12 months after
transplant [32]. Firstly, levels of memory followed by transitional and
naïve B cells rise [33,34]. Recovery of B lymphocytes in children has
been observed to be significantly faster after unrelated transplant of
cord blood than BMT, although without impact on overall survival
[35]. PBSCT results in higher counts of B lymphocytes in the
peripheral blood early after engraftment compared to BMT, whereas
there is no difference later [36]. Peripheral blood B cells don't reach
normal counts until up to one year after HSCT and consist of only
donor-derived lymphocytes [30,37]. GvHD is an important factor
in delayed reconstitution of lymphocyte pools, and especially B
cells after HSCT [27,32,38]. Furthermore, development of chronic
GvHD correlates with delayed recovery of B lymphocytes [32].
Concomitantly, high counts of B cell precursors correlate with a
significantly lower incidence of cGvHD, indicating unaltered B cell
lymphopoiesis [39]. Increased numbers of transitional [37] and naïve
B cells [40] are found after HSCT. Early after transplant, the B cell
CDR3 repertoire is restricted, but after 12 months post-HSCT the
variability of naïve B lymphocyte pattern isn't significantly different
from healthy controls, whereas a restricted repertoire of memory B
cells still exists [41]. Up to 6 months after HSCT, IgH CD3 repertoire
disparities between memory and naïve B cells have been detected,
indicating compromised reconstitution of CD27+ memory B
lymphocytes [37,40,42]. In pediatric patients, CD27- IgMhigh cells have
been observed to constitute the largest population of B lymphocytes in
the first 12 months after HSCT, with levels decreasing over time. This
subset has been further divided into transitional and non-transitional
cells, which in turn were differentiated via CD45RBMEM55 [31]. In the
first two years, levels of memory B cell are very low whereas naïve
B cells are predominant and B lymphocyte reconstitution resembles
the physiologic development in children [27]. Furthermore, the
level of CD27+ IgM- memory cells has been found to proportionally
increase, whereas CD27+ IgMhigh memory B cells would not increase
and stay lower than compared to a control group [31]. It has been
proposed, that the occurrence of new B cells after HSCT may serve as
a predictive determinant of later B cell levels [43].
Bcells and cGvHD
In models for several autoimmune diseases, including rheumatic
arthritis and JIA,autoreactive B cells, emerging from failure of
tolerance checkpoints, play an important role [1,44]. Elevated levels
of TNFα and IL-6, commonly associated with classic autoimmune
diseases, can also be found in cGvHD patients [11]. Delayed or
disturbed B cell-reconstitution and elevated plasma B cell–activating
factor levels are associated with increased numbers of circulating
CD27+ B cell subsets [45]. Furthermore, this observation is associated
with significantly higher B cell-protein contents [46], which comprise
antigen-experienced B cells with a correlating commitment to plasma
cell differentiation [47]. Indeed did we find higher levels of memory
B cells in the patient population as compared to healthy donors. In
addition, cGvHD patients who demonstrate clinical improvement
and positive response to treatment have robust recovery of the
peripheral naïve B cell pool [45,48]. This suggests, concomitant with
an observed increase of IgD+ CD38high CD27- in healthy HSCT patients
[45], that the return to B cell homeostasis might be critical in the
prevention of autoimmune cGvHD [49] and indeed, in our patients
we observed less naïve IgD+ CD27- B cells, as well as increased levels
of transitional cells. This might be due to delayed and impaired B cell
reconstitution in cGvHD patients. CD34- CD20+ B cells, known to
positively correlate, where as CD34+ CD19+ progenitor B cells in the
transplant would correlate inversely with GvHD incidence [50]. In
patients with cGVHD, less CD24hi CD27+ BcellsandIL-10–producing
CD24hi CD27+ Bcells have been detected [42]. Nevertheless, after HSCT
no significant correlation between B cell subsets in the bone marrow
and cGvHD or survival could be detected [51]. With the knowledge,
that induction of cGvHD depends on interaction of CD4+ T cells with
B cells, it surprises, that the absence of B cells in secondary recipients
didn't prevent cGvHD after CD4+ T cells already had contact with
B cells [13]. Auto antibodies found in patients with cGvHD are of
various types, including ANA, anti-dsDNA, anti-smooth muscle, anti
mitochondrial and anti cardiolipin antibodies [52,53]. In patients
with extensive GvHD, auto antibodies specifically targeting PDGFR
and mediating excessive production of collagen have been identified,
their levels matching the severity of skin involvement or lung fibrosis
[16]. The occurrence of auto antibodies could be associated with a
higher incidence of cGvHD and a lower relapse rate [53]. However,
even if studies find correlations of antibodies with disease severity
[54], their occurrence is inconsistent [55]. In addition to the described
interactions of B cell subsets and cGvHD, we detected significantly
higher levels of CD69+ and CD80+ B cells in patients as compared
to healthy donors, indicating higher levels of activation. Activated
alloreactive and autoreactive B cells are associated with cGvHD [56].
With no significant differences in B cell subsets concerning apoptosis,
our co-cultivation experiments do not support the observations made
in naïve and non-switched memory B cells.
DCs and cGvHD
A decrease in GvHD severity after depletion of CD11chigh donor
DCs has been observed [57]. In addition, different manifestations
seem to be dependent on distinctive APC profiles as, for example,
expression of CD80/86 in T cells for induction of cutaneous cGvHD,
which could be mediated by both donor and host APCs, whereas
gut cGvHD was mainly dependent on donor APCs and required
both CD80/86 and CD40 signaling [58]. CD83, highly expressed
on DCs, is suggested to contribute to pathways including activation
of CD4+ and CD8+ T cells as well as B cell homeostasis [59]. PDCs
are characterized by a CD11cint B220+ phenotype and are capable
of producing high amounts of type I IFN as well as exerting potent
T cell stimulation when matured via CD40 or TLR [25,60,61]. This
stresses the importance of donor DCs, as 80% of peripheral blood
DCs are replaced by donor cells within 14 days after transplantation
in human recipients, reaching more than 95% by day 56 [62].
Reconstitution of DCs starts at about 2 to 3 weeks after HSCT, with
numbers of myeloid DCs normalizing, while pDCs don't reach
normal levels even in 12 months [27]. No significant association
between DC chimerism and cGvHD has yet been found [63], but
there is evidence, that host DCs can present host-derived antigen to
donor T cells and donor DCs have activating effects on alloreactive
CD8+ T cells [64]. Nevertheless, a profound depletion of host DCs
and B cells could not prevent GvHD induction [65]. Donor CD11b-
CD11c+ pDCs have been observed to enhance Th1, CD4+ and type I
CD8+ CTL immune polarization of donor T cells as well as promoting
GVL effects without enhancing GvHD, while CD11b+ CD11c+ cDCs
would induce Th2 and type II CD8+ CTL immune polarization
[66]. This complements the observation that adding donor myeloid
or CD11chigh pDCs to the hematopoietic graft results in increased
severity of GvHD [67]. Our co-cultivation experiments reveal
significant differences in CD11c- DCs only between healthy donors
and auto-allo, indicating a mitigating effect of patient B cells on donor
DC homeostasis. Experimental depletion of host CD11+ DCs wasn't
able to prevent the disease [65]. Instead, transplantation of CD11b
depleted bone marrow hematopoietic grafts leads to increased levels
of donor spleen-derived CD4+ memory T cells mediating augmented
GVL effects and IFNγ in the recipient [68]. However, higher counts
of CD123+ CD4+ DCs in the bone marrow graft have been found
to be associated with decreased incidence of cGvHD but increased
relapse in recipients [69]. Lack of CD4+ CD25+ Foxp3+ Tregs can
induce severe autoimmunity [70]. In the presence of TGFβ and alltrans
retinoic acid, mouse DCs can indeed induce antigen-specific
and immunosuppressive CD4+ CD25+ Foxp3+ Tregs capable of
persisting for months and attenuating GvHD [71]. Interestingly, in
patients with cGvHD, Th17 and CD4+ CD25+ Foxp3+ T cells exhibit
an inverse proportionality [11]. Levels of Th17 cells in patients with
active cGvHD are significantly elevated in comparison to healthy
donors; furthermore, patients with inactive GvHD have very low
counts of Th17 cells in the peripheral blood, making these cells good
indicators for disease status [11]. In mouse models, treatment with
regulatory DCs after allogeneic HSCT can mediate an increase in
levels of antigen-specific CD4+ CD25+ Foxp3+ T cells and thus prevent
cutaneous cGvHD in mice by generating alloreactive Tregs from
donor derived T cells [72]. Furthermore, experimental therapeutic
extracorporeal photopheresis increased levels of CD4+ CD25+ Foxp3+
and modulate cGvHD activity [73]. Yet, the genotype of the donor
for the (GT)n Polymorphism in the Promoter/Enhancer of Focp3 has
no correlation with the development of cGvHD [74]. Type I IFN acts
in autoimmune pathogenesis by negatively affecting differentiation
of Th2 and Th17 cells [64,75] and inducing antibody responses [26].
The immuno modulatory effects of type I IFN include increased
maturation of DCs and upregulation of expression of MHC I and
II, as well as enhanced levels of CD80/86, BAFF and APRIL [25]. In
patients after allogeneic HSCT, CD4+ CD123+ pDC precursors were
able to produce type I IFN [76] and in mice, pDCs expressing MHCII
are able to sufficiently prime donor CD4+ T cells to induce GvHD
[61]. On the other hand, secretion of IFNγ by murine donor T cells
resulted in increased expression of Indoleamine-2,3-dioxygenase,
involved in the regulation of gastrointestinal GvHD, [77] in donor
pDCs, thus altering T cell balance and limiting GvHD without
inhibition of GVL effects [78]. Furthermore, high levels of IL-15 on
day +7 post transplant have been observed to correlate significantly
with a lower risk of cGvHD [79], being involved in autoimmune
diseases as a modulator of inflammation [80] and enhancing GVL
effects [81]. We observed no effects of DCs on B-cell-dependent
pathways, possibly due to less CD11c-regulatory DCs and high levels
of apoptosis.
Biomarkers
Besides a disturbed B cell-homeostasis [49,51], some previously
described biomarkers have been associated with an involvement of
specific organs, such as soluble CD13 and BAFF for hepatic, and antidsDNA
antibodies for joint, sclerodermatous and ocular cGvHD
[52]. In pediatric patients, high levels of IL-2Rα and HGF could be
significantly associated with GvHD, whereas elevated IL-8 would
decrease the risk of GvHD [82]. Concentrations of CXCL9 above the
median have been found to be associated with cGvHD within the first
three months of diagnosis [83]. CXCL10 and CXCL11 have lately
been identified as promising diagnostic markers for both aGvHD
and cGvHD [84]. BAFF also has been suggested as a biomarker for
cGvHD [49,85], it acts as a survival factor for B cells, as high levels of
BAFF have been shown to result in increased size and metabolism of
B cells [46]. Elevated levels of BAFF in patients with cGvHD positively
affect numbers of both, pre-germinal center and post-germinal center
plasmablast- and plasmacell-like B cell subsets [45]. In transgenic
mice models, over expression of BAFF prevents apoptosis of selfreactive
B cells, leads to an enhancement of anti-dsDNA-specific B
cell-maturation as well as secretion of antibodies [86] and results in
autoimmune phenomena [87]. Interestingly, BAFF-receptor deficient
mice would not develop autoimmunity, indicating a prominent
role of this factor in disease pathogenesis [88]. Levels of BAFF are
significantly higher in patients with clinical manifestation of cGvHD
than in patients without cGvHD [89]. An elevated BAFF/B cell-ratio
in patients with cGvHD has been observed due to both persistent
elevation of BAFF concentration and B-lymphopenia, characterized
by delayed recovery of B cell homeostasis and a selective defect in
numbers of naive CD27- cells [45]. Despite that, a decrease of soluble
BAFF levels could predict the clinical response to cGvHD [52]. The
possibility of BAFF-mediated activation of AKT and ERK pathways
in patients with cGvHD has been proposed, resulting in decreased B
cell-apoptosis by lower levels of anti apoptotic Bim via NF-κB pathway
[46,90]. IL-10 is an important factor in autoimmune pathogenesis
as observed in experimental autoimmune encephalomyelitis,
inflammatory bowel disease, collagen-induced arthritis and systemic
lupus erythematosus [91]. Maturation of DCs under the influence of
IL-10 induces tolerogenic DCs capable of inducing anergic CD4+ and
CD8+ cells, inhibiting Th1 and Th2 responses [60]. B cells expressing
B7 are necessary to mediate expression of IL-10 and Foxp3 by
CD4+ CD25+ Treg cells in experimental autoimmune encephalitis,
thus supporting recovery from the disease [92]. In mouse models,
transferred IL-10-producing B cells were able to reduce cGvHD
severity due to reconstitution of regulatory B cell subsets and
have been shown to have a suppressive role in the development of
sclerodermatous cGvHD [93], as well as mediating the decrease of
allogeneic donor T cell proliferation and expansion [94]. In patients
with cGvHD, an impaired production of IL-10 has been observed
[42]. The detection of allogeneic HY-antibodies 3 months after female
to male HSCT may predict incidence and severity of cGvHD as well
as non relapse mortality [95].
Approaches to targeted therapy
Present B cell targeted therapies include anti-CD20, -CD22, -IL-
12/23, -BAFF and -BAFF-receptor-3 antibodies [1,96-98]. A series
of studies have analyzed the use of rituximab in human cGvHD
with response rates ranging from 50 to 83% [54]. After treatment
with rituximab, levels of precursor B cells were significantly higher
in patients with stable or improved disease showing the correlation
between clinical response and B cell-homeostasis [99]. On the other
hand, it has been suggested that rituximab, failing to reduce BAFF
levels, would induce an even more abnormal BAFF/B cell-ratio and
support activated B cells [45] and its application as a prophylactic
agent is still controversially discussed [18,99,100]. Dendritic
vaccination is a promising approach to induce direct tumor-targeted
immune responses after HSCT in order to enhance GVL effects.
Ideally, immune responses against the malignant cells mediated
by antigen-specific DCs would not have effects on cGvHD [101].
However, GvHD negatively affects the desired reactions to vaccines
[102]. Nonetheless, the setting of HSCT is ideal for application of
DCs, because, as mentioned earlier, donor DCs tend to fully replace
host DCs after transplantation [62]. There are several approaches
to dendritic vaccination against neoplasms, including generation of
DCs ex vivo by culturing progenitor cells with specific cytokines and
infusion of these DCs in patients, tumor-associated antigens aimed at
DCs, and DC-derived exosomes [19]. Disturbed B cell-homeostasis
and increased autoreactive B cells–critical in the development of
cGvHD [103] are mediated, amongst others, by type I IFN-producing
DCs and increased BAFF levels. Both could be influenced by in-vitro
stimulated pDCs. Further innovative approaches in GvHD therapy
include tyrosine kinase inhibitors like Imatinibvia Inhibition of
c-Abl activity possibly blocking autoreactive B cell responses [104]
and Ibrutinib, an inhibitor of both Bruton’s Tyrosine Kinase and
IL-2 Inducible T-cell Kinase, enzymes upstream of BCR and TCR
signaling pathways, having shown promising results in acting as a
prophylactic agent in murine cGvHD models [105].
Table 3
Table 3
Conclusions and Perspectives
Summarizing these observations, an impaired reconstitution of the immune cell homeostasis, in particular B cells, seems to be the basis of cGvHD pathogenesis, characterized by an increased memory, transitional, CD69+ and CD86+ phenotype and lower levels of naïve B cells due to Apoptosis.However, despite extensive investigations, there is little evidence that findings have facilitated a cohesive understanding, have translated into clinically useful tools, or have induced new therapeutic strategies. Validated markers for the risk of cGvHD development may permit avoidance of donors prone to cause cGvHD, alteration of transplant or therapeutic approaches to mitigate these risks. Useful and validated biologic markers associated with treatment-response and prognosis might positively influence current clinical practice and augment the range of therapeutic intervention. An advanced understanding of genetic polymorphisms in several cytokines, donor-recipient HLA-matching, MHC haplotypes and minor histocompatibility mismatch [106] may help to understand pathophysiological development of cGvHD [107,108]. As various interactions between B cells and DCs exist, DC-mediated priming of B cells or dendritic vaccinations seem to imply new therapeutic possibilities, even if our results dispute a role of DCs in altered B cell homeostasis in cGvHD patients. A priority area for future research lies in well-characterized clinical populations in order to obtain adequate power for studies in this field. Translation of experimental data into a clinical setting might provide useful tools for assessing risk for cGvHD and response, and may identify novel therapeutic strategies to reduce morbidity and improve prognosis.
Acknowledgement
This publication was supported by the Open Access Publication Fund of the University of Wuerzburg.
References
- Shimabukuro-Vornhagen A, Hallek MJ, Storb RF, Bergwelt-Baildon MS von. The role of B cells in the pathogenesis of graft-versus-host disease. Blood. 2009; 114: 4919-4927.
- Oberg JA, Bender JG, Morris E, Harrison L, Basch CE, Garvin JH, et al. Pediatric allo-SCT for malignant and non-malignant diseases: impact on health-related quality of life outcomes. Bone Marrow Transplant. 2013; 48: 787-793.
- Storb R, Gyurkocza B, Storer BE, Sorror ML, Blume K, Niederwieser D, et al. Graft-Versus-Host Disease and Graft-Versus-Tumor Effects After Allogeneic Hematopoietic Cell Transplantation. J Clin Oncol. 2013; 31: 1530-1538.
- Boyiadzis M, Arora M, Klein JP, Hassebroek A, Hemmer M, Urbano-Ispizua A, et al. Impact of Chronic Graft-versus-Host Disease on Late Relapse and Survival on 7,489 Patients after Myeloablative Allogeneic Hematopoietic Cell Transplantation for Leukemia. Clin cancer Res. 2015; 21: 2020-2028.
- Zinöcker S, Dressel R, Wang X-N, Dickinson AM, Rolstad B. Immune reconstitution and graft-versus-host reactions in rat models of allogeneic hematopoietic cell transplantation. Front. Immunol. 2012; 3: 355.
- Baird K, Cooke K, Schultz KR. Chronic Graft-Versus-Host Disease (GVHD) in Children. Pediatr Clin North Am. 2010; 57: 297-322.
- Eapen M, Horowitz MM, Klein JP, Champlin RE, Loberiza FR, Ringdén O, et al. Higher mortality after allogeneic peripheral-blood transplantation compared with bone marrow in children and adolescents: the Histocompatibility and Alternate Stem Cell Source Working Committee of the International Bone Marrow Transplant Registry. J Clin Oncol. 2004; 22: 4872-4880.
- Miklos DB, Kim HT, Miller KH, Guo L, Zorn E, Lee SJ, et al. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood. 2005; 105: 2973-2978.
- Welniak LA, Blazar BR, Murphy WJ. Immunobiology of allogeneic hematopoietic stem cell transplantation. Annu. Rev. Immunol. 2007; 25: 139-170.
- Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol 2012; 12: 443-458.
- Dander E, Balduzzi A, Zappa G, Lucchini G, Perseghin P, Andrè V, et al. Interleukin-17-producing T-helper cells as new potential player mediating graft-versus-host disease in patients undergoing allogeneic stem-cell transplantation. Transplantation. 2009; 88: 1261-1272.
- Mitsdoerffer M, Lee Y, Jäger A, Kim H-J, Korn T, Kolls JK et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci USA. 2010; 107: 14292-14297.
- Young JS, Wu T, Chen Y, Zhao D, Liu H, Yi T, et al. Donor B cells in transplants augment clonal expansion and survival of pathogenic CD4+ T cells that mediate autoimmune-like chronic graft-versus-host disease. J Immunol. 2012; 189: 222-233.
- Ferrara JLM, Levine JE, Reddy P, Holler E. Graft-versus-host disease. The Lancet. 2009; 373: 1550-1561.
- Zhang C, Todorov I, Zhang Z, Liu Y, Kandeel F, Forman S, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood. 2006; 107: 2993-3001.
- Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, Trappolini S, et al. Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood. 2007; 110: 237-241.
- Paczesny S. Discovery and validation of graft-versus-host disease biomarkers. Blood. 2013; 121: 585-594.
- Arai S, Sahaf B, Narasimhan B, Chen GL, Jones CD, Lowsky R, et al. Prophylactic rituximab after allogeneic transplantation decreases B-cell alloimmunity with low chronic GVHD incidence. Blood. 2012; 119: 6145-6154.
- Galluzzi L, Senovilla L, Vacchelli E, Eggermont A, Fridman WH, Galon J, et al. Trial watch: Dendritic cell-based interventions for cancer therapy. Oncoimmunology. 2012; 1: 1111-1134.
- Ding C, Cai Y, Marroquin J, Ildstad ST, Yan J. Plasmacytoid Dendritic Cells Regulate Autoreactive B Cell Activation via Soluble Factors and in a Cell-to-Cell Contact Manner. J Immunol. 2009; 183: 7140-7149.
- Kleindienst P, Brocker T. Concerted antigen presentation by dendritic cells and B cells is necessary for optimal CD4 T-cell immunity in vivo. Immunology 2005; 115: 556-564.
- Jego G, Palucka AK, Blanck J-P, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003; 19: 225-234.
- Gilliet M, Cao W, Liu Y-J. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008; 8: 594-606.
- Shaw J, Wang Y-H, Ito T, Arima K, Liu Y-J. Plasmacytoid dendritic cells regulate B-cell growth and differentiation via CD70. Blood. 2010; 115: 3051-3057.
- Rönnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008; 17: 394-399.
- Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol. 2006; 176: 2074-2078.
- Storek J, Geddes M, Khan F, Huard B, Helg C, Chalandon Y, et al. Reconstitution of the immune system after hematopoietic stem cell transplantation in humans. Semin Immunopathol. 2008; 30: 425-437.
- Douagi I, Gujer C, Sundling C, Adams WC, Smed-Sorensen A, Seder RA, et al. Human B Cell Responses to TLR Ligands Are Differentially Modulated by Myeloid and Plasmacytoid Dendritic Cells. J Immunol. 2009; 182: 1991-2001.
- Guan X, Peng J-R, Yuan L, Wang H, Wei Y-H, Leng X-S. A novel, rapid strategy to form dendritomas from human dendritic cells and hepatocellular carcinoma cell line HCCLM3 cells using mature dendritic cells derived from human peripheral blood CD14+ monocytes within 48 hours of in vitro culture. World journal of gastroenterology: WJG 2004; 10: 3564-3568.
- Bosch M, Khan FM, Storek J. Immune reconstitution after hematopoietic cell transplantation. Curr Opin Hematol. 2012; 19: 324-335.
- Bemark M, Friskopp L, Saghafian-Hedengren S, Koethe S, Fasth A, Abrahamsson J, et al. A glycosylation-dependent CD45RB epitope defines previously unacknowledged CD27−IgMhigh B cell subpopulations enriched in young children and after hematopoietic stem cell transplantation. Clinical Immunology. 2013; 149: 421-431.
- Bae KW, Kim BE, Koh KN, Im HJ, Seo JJ. Factors influencing lymphocyte reconstitution after allogeneic hematopoietic stem cell transplantation in children. Korean J Hematol. 2012; 47: 44-52.
- Mensen A, Ochs C, Stroux A, Wittenbecher F, Szyska M, Imberti L, et al. Utilization of TREC and KREC quantification for the monitoring of early T- and B-cell neogenesis in adult patients after allogeneic hematopoietic stem cell transplantation. J Transl Med. 2013; 11: 188.
- Federmann B, Hägele M, Pfeiffer M, Wirths S, Schumm M, Faul C, et al. Immune reconstitution after haploidentical hematopoietic cell transplantation: impact of reduced intensity conditioning and CD3/CD19 depleted grafts. Leukemia. 2010; 25: 121-129.
- Rénard C, Barlogis V, Mialou V, Galambrun C, Bernoux D, Goutagny MP, et al. Lymphocyte subset reconstitution after unrelated cord blood or bone marrow transplantation in children. Br J Haematol. 2011; 152: 322-330.
- Storek J. Immune reconstitution after allogeneic marrow transplantation compared with blood stem cell transplantation. Blood. 2001; 97: 3380-3389.
- Bemark M, Holmqvist J, Abrahamsson J, Mellgren K. Translational Mini-Review Series on B cell subsets in disease. Reconstitution after haematopoietic stem cell transplantation - revelation of B cell developmental pathways and lineage phenotypes. Clin Exp Immunol. 2012; 167: 15-25.
- Olkinuora H, Willebrand E von, Kantele JM, Vainio O, Talvensaari K, Saarinen-Pihkala U, et al. The Impact of Early Viral Infections and Graft-Versus-Host Disease on Immune Reconstitution Following Paediatric Stem Cell Transplantation. Scand J Immunol. 2011; 73: 586-593.
- Fedoriw Y, Samulski TD, Deal AM, Dunphy CH, Sharf A, Shea TC, et al. Bone Marrow B cell Precursor Number after Allogeneic Stem Cell Transplantation and GVHD Development. Biol Blood Marrow Transplant. 2012; 18: 968-973.
- Avanzini MA, Locatelli F, Dos Santos C, Maccario R, Lenta E, Oliveri M, et al. B lymphocyte reconstitution after hematopoietic stem cell transplantation: functional immaturity and slow recovery of memory CD27+ B cells. Exp. Hematol. 2005; 33: 480-486.
- Omazic B, Lundkvist I, Mattsson J, Permert J, Nasman-Bjork I. Memory B lymphocytes determine repertoire oligoclonality early after haematopoietic stem cell transplantation. Clin Exp Immunol. 2003; 134: 159-166.
- Masson A de, Bouaziz J-D, Le Buanec H, Robin M, O'Meara A, Parquet N, et al. CD 24(hi)CD27⁺ and plasmablast-like regulatory B cells in human chronic graft-versus-host disease. Blood. 2015; 125: 1830-1839.
- Serana F, Sottini A, Chiarini M, Zanotti C, Ghidini C, Lanfranchi A, et al. The Different Extent of B and T Cell Immune Reconstitution after Hematopoietic Stem Cell Transplantation and Enzyme Replacement Therapies in SCID Patients with Adenosine Deaminase Deficiency. J Immunol. 2010; 185: 7713-7722.
- Wiegering V, Girschick HJ, Morbach H. B-cell pathology in juvenile idiopathic arthritis. Arthritis. 2010; 2010: 759868.
- Sarantopoulos S, Stevenson KE, Kim HT, Cutler CS, Bhuiya NS, Schowalter M, et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood. 2009; 113: 3865-3874.
- Allen JL, Fore MS, Wooten J, Roehrs PA, Bhuiya NS, Hoffert T, et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood. 2012; 120: 2529-2536.
- Avery DT, Ellyard JI, Mackay F, Corcoran LM, Hodgkin PD, Tangye SG. Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. J Immunol. 2005; 174: 4034-4042.
- Kuzmina Z, Greinix HT, Weigl R, Körmöczi U, Rottal A, Frantal S, et al. Significant differences in B-cell subpopulations characterize patients with chronic graft-versus-host disease-associated dysgammaglobulinemia. Blood. 2011; 117: 2265-2274.
- Levine JE, Paczesny S, Sarantopoulos S. Clinical applications for biomarkers of acute and chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2012; 18: 24.
- Iori AP, Torelli GF, Propris MS de, Milano F, Pupella S, Gozzer M, et al. B-cell concentration in the apheretic product predicts acute graft-versus-host disease and treatment-related mortality of allogeneic peripheral blood stem cell transplantation. Transplantation. 2008; 85: 386-390.
- Michonneau D, Peffault de Latour R, Porcher R, Robin M, Benbunan M, Rocha V, et al. Influence of bone marrow graft B lymphocyte subsets on outcome after HLA-identical sibling transplants. Br J Haematol. 2009; 145: 107-114.
- Fujii H, Cuvelier G, She K, Aslanian S, Shimizu H, Kariminia A, et al. Biomarkers in newly diagnosed pediatric-extensive chronic graft-versus-host disease: a report from the Children's Oncology Group. Blood. 2008; 111: 3276-3285.
- Moon J-H, Lee S-J, Kim J-G, Chae Y-S, Kim S-N, Kang B-W, et al. Clinical significance of autoantibody expression in allogeneic stem-cell recipients. Transplantation. 2009; 88: 242-250.
- Kapur R, Ebeling S, Hagenbeek A. B-cell involvement in chronic graft-versus-host disease. Haematologica. 2008; 93: 1702-1711.
- Socie G. Chronic GVHD: B cells come of age. Blood. 2011; 117: 2086-2087.
- Socié G, Ritz J. Current issues in chronic graft-versus-host disease. Blood. 2014; 124: 374-384.
- MacDonald KP, Kuns RD, Rowe V, Morris ES, Banovic T, Bofinger H, et al. Effector and regulatory T-cell function is differentially regulated by RelB within antigen-presenting cells during GVHD. Blood. 2007; 109: 5049-5057.
- Anderson BE, McNiff JM, Jain D, Blazar BR, Shlomchik WD, Shlomchik MJ. Distinct roles for donor- and host-derived antigen-presenting cells and costimulatory molecules in murine chronic graft-versus-host disease: requirements depend on target organ. Blood. 2005; 105: 2227-2234.
- Ehlers C, Schirmer S, Kehlenbach RH, Hauber J, Chemnitz J. Post-transcriptional regulation of CD83 expression by AUF1 proteins. Nucleic Acids Res. 2013; 41: 206-219.
- Hubo M, Trinschek B, Kryczanowsky F, Tuettenberg A, Steinbrink K, Jonuleit H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front Immunol. 2013; 4: 82.
- Koyama M, Hashimoto D, Aoyama K, Matsuoka K-I, Karube K, Niiro H, et al. Plasmacytoid dendritic cells prime alloreactive T cells to mediate graft-versus-host disease as antigen-presenting cells. Blood. 2009; 113: 2088-2095.
- Auffermann-Gretzinger S, Lossos IS, Vayntrub TA, Leong W, Grumet FC, Blume KG, et al. Rapid establishment of dendritic cell chimerism in allogeneic hematopoietic cell transplant recipients. Blood. 2002; 99: 1442-1448.
- Pihusch M, Boeck S, Hamann M, Pihusch V, Heller T, Diem H, et al. Peripheral dendritic cell chimerism in allogeneic hematopoietic stem cell recipients. Transplantation. 2005; 80: 843-849.
- Charrier E, Cordeiro P, Cordeau M, Dardari R, Michaud A, Harnois M, et al. Post-transcriptional down-regulation of Toll-like receptor signaling pathway in umbilical cord blood plasmacytoid dendritic cells. Cell Immunol. 2012; 276: 114-121.
- Li H, Demetris AJ, McNiff J, Matte-Martone C, Tan HS, Rothstein DM, et al. Profound Depletion of Host Conventional Dendritic Cells, Plasmacytoid Dendritic Cells, and B Cells Does Not Prevent Graft-versus-Host Disease Induction. J Immunol. 2012; 188: 3804-3811.
- Li J-M, Southerland LT, Lu Y, Darlak KA, Giver CR, McMillin DW, et al. Activation, immune polarization, and graft-versus-leukemia activity of donor T cells are regulated by specific subsets of donor bone marrow antigen-presenting cells in allogeneic hemopoietic stem cell transplantation. J Immunol. 2009; 183: 7799-7809.
- MacDonald KPA, Rowe V, Clouston AD, Welply JK, Kuns RD, Ferrara JLM, et al. Cytokine expanded myeloid precursors function as regulatory antigen-presenting cells and promote tolerance through IL-10-producing regulatory T cells. J Immunol. 2005; 174: 1841-1850.
- Li J-M, Waller EK. Donor antigen-presenting cells regulate T-cell expansion and antitumor activity after allogeneic bone marrow transplantation. Biol Blood Marrow Transplant. 2004; 10: 540-551.
- Waller EK, Rosenthal H, Jones TW, Peel J, Lonial S, Langston A, et al. Larger numbers of CD4(bright) dendritic cells in donor bone marrow are associated with increased relapse after allogeneic bone marrow transplantation. Blood. 2001; 97: 2948-2956.
- Buckner JH. Mechanisms of impaired regulation by CD4+CD25+FOXP3+ regulatory T cells in human autoimmune diseases. Nat Rev Immunol 2010; 10: 849-859.
- Sela U, Olds P, Park A, Schlesinger SJ, Steinman RM. Dendritic cells induce antigen-specific regulatory T cells that prevent graft versus host disease and persist in mice. J Exp Med. 2011; 208: 2489-2496.
- Fujita S, Sato Y, Sato K, Eizumi K, Fukaya T, Kubo M, et al. Regulatory dendritic cells protect against cutaneous chronic graft-versus-host disease mediated through CD4+CD25+Foxp3+ regulatory T cells. Blood. 2007; 110: 3793-3803.
- Capitini CM, Davis JP, Larabee SM, Herby S, Nasholm NM, Fry TJ. Extracorporeal Photopheresis Attenuates Murine Graft-versus-Host Disease via Bone Marrow–Derived Interleukin-10 and Preserves Responses to Dendritic Cell Vaccination. Biology of Blood and Marrow Transplantation. 2011; 17: 790-799.
- Noriega V, Martínez-Laperche C, Buces E, Pion M, Sánchez-Hernández N, Martín-Antonio B, et al. The Genotype of the Donor for the (GT)n Polymorphism in the Promoter/Enhancer of FOXP3 Is Associated with the Development of Severe Acute GVHD but Does Not Affect the GVL Effect after Myeloablative HLA-Identical Allogeneic Stem Cell Transplantation. PloS one. 2015; 10: e0140454.
- Huber JP, Farrar JD. Regulation of effector and memory T-cell functions by type I interferon. Immunology. 2011; 132: 466-474.
- Fagnoni FF, Oliviero B, Giorgiani G, Stefano P de, Dehò A, Zibera C, et al. Reconstitution dynamics of plasmacytoid and myeloid dendritic cell precursors after allogeneic myeloablative hematopoietic stem cell transplantation. Blood. 2004; 104: 281-289.
- Ratajczak P, Janin A, Peffault de Larour R, Koch L, Roche B, Munn D, et al. IDO in human gut graft-versus-host disease. Biol Blood Marrow Transplant. 2012; 18: 150-155.
- Lu Y, Giver CR, Sharma A, Li JM, Darlak KA, Owens LM, et al. IFN-γ and indoleamine 2,3-dioxygenase signaling between donor dendritic cells and T cells regulates graft versus host and graft versus leukemia activity. Blood. 2012; 119: 1075-1085.
- Pratt LM, Liu Y, Ugarte-Torres A, Hoegh-Petersen M, Podgorny PJ, Lyon AW, et al. IL15 levels on day 7 after hematopoietic cell transplantation predict chronic GVHD. Bone Marrow Transplant. 2013; 48: 722-728.
- Ohteki T, Tada H, Ishida K, Sato T, Maki C, Yamada T, et al. Essential roles of DC-derived IL-15 as a mediator of inflammatory responses in vivo. J Exp Med. 2006; 203: 2329-2338.
- Sauter CT, Bailey CP, Panis MM, Biswas CS, Budak-Alpdogan T, Durham A, et al. Interleukin-15 administration increases graft-versus-tumor activity in recipients of haploidentical hematopoietic SCT. Bone Marrow Transplant. 2013; 48: 1237-1242.
- Berger M, Signorino E, Muraro M, Quarello P, Biasin E, Nesi F et al. Monitoring of TNFR1, IL-2Rα, HGF, CCL8, IL-8 and IL-12p70 following HSCT and their role as GVHD biomarkers in paediatric patients. Bone Marrow Transplant. 2013; 48: 1230-1236.
- Kitko CL, Levine JE, Storer BE, Chai X, Fox DA, Braun TM, et al. Plasma CXCL9 elevations correlate with chronic GVHD diagnosis. Blood. 2014; 123: 786-793.
- Ahmed SS, Wang XN, Norden J, Pearce K, El-Gezawy E, Atarod S, et al. Identification and validation of biomarkers associated with acute and chronic graft versus host disease. Bone marrow transplant. 2015; 50: 1563-1571.
- Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/APRIL system: Emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013; 24: 203-215.
- Thorn M, Lewis RH, Mumbey-Wafula A, Kantrowitz S, Spatz LA. BAFF overexpression promotes anti-dsDNA B-cell maturation and antibody secretion. Cell Immunol. 2010; 261: 9-22.
- Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004; 20: 785-798.
- Mayne CG, Nashold FE, Sasaki Y, Hayes CE. Altered BAFF-receptor signaling and additional modifier loci contribute to systemic autoimmunity in A/WySnJ mice. Eur J Immunol. 2009; 39: 589-599.
- Kuzmina Z, Krenn K, Petkov V, Körmöczi U, Weigl R, Rottal A, et al. CD19(+)CD21(low) B cells and patients at risk for NIH-defined chronic graft-versus-host disease with bronchiolitis obliterans syndrome. Blood. 2013; 121: 1886-1895.
- Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont M-C, Ranchin B, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009; 10: 778-785.
- Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol. 2008; 8: 391-397.
- Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B cell regulation of CD4+CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol. 2007; 178: 3447-3456.
- Le Huu D, Matsushita T, Jin G, Hamaguchi Y, Hasegawa M, Takehara K, et al. Donor-derived regulatory B cells are important for suppression of murine sclerodermatous chronic graft-versus-host disease. Blood. 2013; 121: 3274-3283.
- Rowe V, Banovic T, MacDonald KP, Kuns R, Don AL, Morris ES, et al. Host B cells produce IL-10 following TBI and attenuate acute GVHD after allogeneic bone marrow transplantation. Blood. 2006; 108: 2485-2492.
- Nakasone H, Tian L, Sahaf B, Kawase T, Schoenrock K, Perloff S, et al. Allogeneic HY antibodies detected 3 months after female-to-male HCT predict chronic GVHD and nonrelapse mortality in humans. Blood. 2015; 125: 3193-3201.
- Sarantopoulos S, Blazar BR, Cutler C, Ritz J. B cells in chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2015; 21: 16-23.
- Okamoto S, Fujiwara H, Nishimori H, Matsuoka K-I, Fujii N, Kondo E, et al. Anti-IL-12/23 p40 antibody attenuates experimental chronic graft-versus-host disease via suppression of IFN-γ/IL-17-producing cells. J Immunol. 2015; 194: 1357-1363.
- Das R, Komorowski R, Hessner MJ, Subramanian H, Huettner CS, Cua D, et al. Blockade of interleukin-23 signaling results in targeted protection of the colon and allows for separation of graft-versus-host and graft-versus-leukemia responses. Blood. 2010; 115: 5249-5258.
- Sarantopoulos S, Stevenson KE, Kim HT, Washel WS, Bhuiya NS, Cutler CS, et al. Recovery of B-cell homeostasis after rituximab in chronic graft-versus-host disease. Blood. 2011; 117: 2275-2283.
- Ratanatharathorn V, Logan B, Wang D, Horowitz M, Uberti JP, Ringden O, et al. Prior rituximab correlates with less acute graft-versus-host disease and better survival in B-cell lymphoma patients who received allogeneic peripheral blood stem cell transplantation. Br J Haematol. 2009; 145: 816-824.
- Toubai T, Sun Y, Luker G, Liu J, Luker KE, Tawara I, et al. Host derived CD8+ dendritic cells are required for induction of optimal graft-versus-tumor responses after experimental allogeneic bone marrow transplantation. Blood. 2013; 121: 4231-4241.
- Capitini CM, Nasholm NM, Duncan BB, Guimond M, Fry TJ. Graft-versus-host disease impairs vaccine responses through decreased CD4+ and CD8+ T cell proliferation and increased perforin-mediated CD8+ T cell apoptosis. J Immunol. 2013; 190: 1351-1359.
- Sarantopoulos S, Ritz J. Aberrant B-cell homeostasis in chronic GVHD. Blood. 2015; 125: 1703-1707.
- Olivieri J, Coluzzi S, Attolico I, Olivieri A. Tirosin kinase inhibitors in chronic graft versus host disease: from bench to bedside. Scientific World Journal. 2011; 11: 1908-1931.
- Schutt SD, Fu J, Nguyen H, Bastian D, Heinrichs J, Wu Y, et al. Inhibition of BTK and ITK with Ibrutinib Is Effective in the Prevention of Chronic Graft-versus-Host Disease in Mice. PloS one. 2015; 10: e0137641.
- Pidala J, Sarwal M, Roedder S, Lee SJ. Biologic markers of chronic GVHD. Bone Marrow Transplant. 2014; 49: 324-331.
- Kohrt HE, Tian L, Li L, Alizadeh AA, Hsieh S, Tibshirani RJ, et al. Identification of gene microarray expression profiles in patients with chronic graft-versus-host disease following allogeneic hematopoietic cell transplantation. Clin Immunol. 2013; 148: 124-135.
- Armistead PM, Liang S, Li H, Lu S, van Bergen CAM, Alatrash G, et al. Common minor histocompatibility antigen discovery based upon patient clinical outcomes and genomic data. PLoS One. 2011; 6: e23217.