Case Report
The Nature of Mutated Muc5ac, an Oncofetal Protein, Expressed in Colorectal Cancer: It’s Role as a Therapeutic Immunogen
Arlen M1*, Arlen P2, Dubeykovskiy A2, Saric O2, Coppa G1, Conte C1 and Molmenti E1
1Department of Surgery, North Shore University Hospital, USA
2Department of Surgery Northwell Health System, Hofstra University College of Medicine, USA
*Corresponding author: Arlen M, Department of Surgery, North Shore University Hospital, USA
Published: 15 Sep, 2016
Cite this article as: Arlen M, Arlen P, Dubeykovskiy A, Saric
O, Coppa G, Conte C, et al. The Nature
of Mutated Muc5ac, an Oncofetal
Protein, Expressed in Colorectal
Cancer: It’s Role as a Therapeutic
Immunogen. Clin Oncol. 2016; 1: 1096.
Abstract
The present concept of effectively treating a malignant lesion, especially one in the process of metastasis, requires being able to turn on cytotoxic T cells that can recognize and destroy the tumor. All attempts at employing this system using TIL and other T cell populations have failed to date. Prehn in the 1950 s postulated that tumors act in a similar fashion to invading bacteria and viruses. Here the host can recognize the foreign invader by defining immunogenic proteins expressed on the cell membrane at the threshold level needed for recognition. He also believed that immunogenic proteins that characterize a malignancy exist, but are only present at a fraction of the level needed to induce tumor destruction. By pooling tumor membrane proteins, we were able to define 3 specific oncofetal proteins expressed only during fetal life. Later in life a viral or carcinogenic agent reactivates the needed gene to produce a post translational modification of the oncofetal protein. The more common of these tumor immunogens to be identified was a modified form of MUC5ac, Monoclonals to this target were produced GMP, tested in vitro and based on the strong ADCC noted in contrast to a CD8 response, were introduced for use in clinical trials in patients with metastatic colorectal cancer having failed all standard forms of therapy.
Introduction
Many have equated the cancer cell to a foreign host invader similar to bacteria and viruses
that enter and infect the host. The cancer cell can also be defined in a similar manner, acting as a
probable host “invader” [1]. The presence a cancer growth results however, in producing greater
consequences. It is far more aggressive, with uncontrolled progression in growth which can lead
to metastasis. Where the malignant cell is genetically programmed to invade and even destroy host
cells in comparison to that of bacteria and viruses, the failure of the existing immune system to
demonstrate any form of immune recognition allows for this process of continued growth of the
tumor [2]. This phenomenon appears to be the result of a weakened tumor immunogenic state.
As such the cancer cell is poorly recognized by host defenses and left to continue in its inherent
growth pattern. In addition, as a means of self protection, the tumor cell does express inhibitory
molecules on its surface which serve to help inhibit the host’s immune system response. [3]. In
order to have proper immunologic recognition of an invading cancer system with the ability of a
patient to reject this type of invader, a threshold level of immunogenic protein that characterizes
a particular malignancy must be presented to the host. In most of the tumor systems that we have
evaluated, a well defined immunogenic oncofetal protein is present, but at such small levels, that the
host is only capable of reaching what is termed a state of “surveillance”. Here an occasional tumor
cell may be destroyed but in general the developing tumor system is left to progress in its pattern
of cellular proliferation. This proliferative state can only be interrupted from an immunologic
standpoint, when an external source of antigen is delivered. This will then bring the threshold level
of tumor immunogen to that level where recognition is accomplished. At this point, the tumor is
then attacked by the host in an attempt to control if not eliminate it. The lack of tumor antigen
expression was first postulated by Prehn [4]. He suggested that while the tumor cells did contain a
low level of immunogen, that the use of pooled allogeneic antigen could possibly, when delivered at
the threshold level, reactivates the needed immune response. He performed various experiment with
mice showing that when the antigen was derived from the pooling of multiple tumor membrane
proteins, that a threshold level could be achieved to prevent tumor growth. Decreasing the level of
antigen resulted in regrowth of tumor. Challenging immunized mice with a different tumor system
failed to prevent tumor growth. The conclusion to such experiments
indicated that vaccines could be effective when delivered at threshold
levels of the needed immunogen, but in addition that the vaccine also
required specificity. In the 1970’s Hollinshead, coming across the
studies provided by Prehn [4] approached the concept of low antigen
expression by tumors, she employed pooled allogeneic membrane
protein to see if improving levels of tumor associated antigen (TAA)
could result in enhancement of the host immune system to target
residual tumor by the host [5]. An initial Phase I study planned by
Hollinshead et al. was performed in the 1980’s. It explored the use
of TAA therapy in patients with several malignancies but in a more
detailed situation with those presenting with adenocarcinoma of
the colon that were demonstrating metastasis or with a very high
possibility of recurrence [6]. The immunogenic tumor proteins
employed in each study were derived from pooled cancer membrane
specimens obtained immediately after surgery [7]. Five years into the
study there was evidence of marked improvement in survival. On that
basis the FDA was approached to allow expansion of such studies
toward developing a commercial product. At that time, the recognition
of AIDS (HIV), Hepatitis C and HPV infections resulted in a change
of procedure under FDA guidance. It was stipulated by FDA, that in
order to advance such trials, a recombinant form of vaccine would
be needed. It was suggested that the protein preparation used in
the initial trials be employed to develop monoclonal antibodies for
affinity purification and to eventually, by mass spectroscopy, define
the structure needed to be sequenced in order to produce the required
recombinant product. Most in our lab considered that contrary to
our initial interpretation of the relative purity of the gel band used
in preparing the final tumor immunogen as defined by patients
being evaluated by delayed cutaneous sensitivity reaction (DHR),
that multiple monoclonals would probably be derived from the
hybridoma preparations that would be developed from the original
antigen preparation. At that point any specific monoclonal antibody
found to target the colon cancer protein would then be used for
affinity purification and sequencing of the antigen to be isolated [8,9].
An HPLC study was performed (Figure 1) utilizing the therapeutic
antigen from the initial clinical trials to develop hybridomas. At least
4 monoclonals were produced, each apparently specific to certain
colorectal carcinoma cells. None reacted with normal colonic tissue.
Three of the mAbs matched the peaks seen on HPLC with that mAb
defining the major peak 4 termed as NPC-1 or Neo 101. The other
mabs studied were labeled as 31.1 and Neo 201. This region (peak
4), comprised the major portion of the antigenic component of the
allogeneic vaccine used in the initial therapeutic trial.
Defining those oncofetal proteins wxpressed in the HPLC
peaks
The murine monoclonal antibody termed mAb NPC-1 (Neo-101)
was directed against and defined that portion of the immunogenic
protein peak 4 seen on the HPLC. This particular protein component
was expressed in more than 60% of the colon cancers examined by
IHC. It proved to be non reactive, and as such absent in normal
adjacent colon tissue [10]. Monoclonal NPC-1C (Chimeric),
developed at Precision Biologics, was found by mass spectroscopy, to
represent that mAb shown to target an altered form of the oncofetal
form of MUC5ac. This mutated protein represented the unique
Tumor Associated Antigen (TAA) comprising a major fraction of
the vaccine that was previously tested in our Phase I clinical colon
trial. The antibodies derived from the hybridomas produced from the
original TAA, were used for further immunopurification and mass
spectroscopy of the several components of the allogeneic vaccine.
The resulting major antigenic component (peak 4) that was defined,
was found to be homologous to the antigen MUC5ac, an oncofetal
protein turned on in the latter part of fetal development [11]. The
NPC-1(Neo-101) monoclonal employed in our studies was developed
at two separate intervals in our lab. The initial Neo -101 was expressed
in the original bioreactor facility at 140 mg. /L of bioreactor fluid
and showed some minimal levels pf toxicity in several patients.
Utilizing the high expression vector developed by Selexis, the new
mAb was now expressed at 2000 mg. /L bioreactor fluid. Studies of
bio similarity were required and revealed that the now termed Neo
-102 was non reactive against red blood cells so that any minor
degree of hemolysis was gone, there was a higher degree of ADCC
and immunohistochemistry (IHC) of tumor specimens expressing
the antigen appeared cleaner. Though both defined similar epitopes,
they were probably glycosylated somewhat differently. This new
version of the mAb targeting the altered form of MUC5ac antigen is
now in a position for commercial development. The NPC-1 antibody
was chimerized by genetically replacing the murine constant regions
with human IgG1 constant regions, and designated asNPC-1C. The
need of course was that for therapeutic purposes, the mechanism for
tumor destruction was through an ADCC (Antibody Dependent Cell
Cytotoxicity) response where the human Fc contained the receptors
for carrying the patients NK cells to the surface of the tumor where
NK destruction of tumor could be initiated. Several laboratory
studies employing fresh-frozen or formalin-fixed tissues indicate
that the NPC-1C antibody stains approximately 60% - 70 % of colon
malignancies. This supported the need for the antigenic protein to be
highly expressed in the tumor cells without cross reacting with normal
tissue. This potential allows the Neo-102 mAb and other similar
monoclonals that we have developed to be used diagnostically, not
only in terms of IHC, but in the development of an accurate serum
ELISA prior to use as a therapeutic [12-14]. (Figure 2) shows the
Optimization Process for use of the mAbs.
Preclinical pharmacology and toxicology studies demonstrated
statistically significant tumor growth inhibition by the mAbs without
significant toxicity, as judged by clinical observation, Studies of body
weights, food consumption, ophthalmology and clinical pathology
in test animals, supported the rationale for development of NPC-
1C antibody for clinical testing since it was the major product
produced by immunization with the tumor antigen. An SDS PAGE
electrophoresis and Western Blot was performed using the NPC-
1 (Neo 102) chimeric of affinity purified NPC-1 antigen (50ugm/
well, lanes 1 and 2) from LS174T lysate (Figure 3). Note that the
high molecular wt. band (approx 600 KD) reacted with Neo-102C
antibody in Western blot. The Coomassie blue band was later excised
and proceeded to MALDI-TOF analysis which revealed its identity as
a MUC5ac modified protein of approximately 600kd.
Figure 1
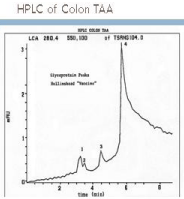
Figure 1
Peak 4 represented what was termed the NPC-1 antigen, that
immunogen comprising a major part of the pooled vaccine protein.
Figure 2
Figure 2
Lead Optimization Process for mAb Products.
Figure 3
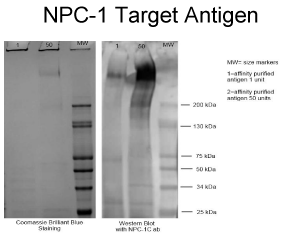
Figure 3
The NPC-1 antigen studied by Western blot and confirmed by
Coomassie Blue staining revealing a molecular band of approximately 600kd.
Background
The origin of the colon oncofetal protein
When the fetus matures in the final term of pregnancy, the gene
needed for producing the oncofetal (immunogenic) protein required
for organ function is demethylase. This allows activation of that
protein or molecule necessary for the fetus to progress to term. In
the case of the colon, the MUC5ac gene is activated to produce the
mucin needed for the bowel and lung to function properly. Just prior
to completion of pregnancy, this gene is re: methylated. It is however
re expressed in some patients later in life where a modification of the
methylated gene by a virus or carcinogen takes place. The oncofetal
protein expressed by this newly modified “malignant cell” has little
resemblance to the original protein functioning in the fetus. During
this post translational state of expression, the modified protein
becomes immunogenic, thus characterizing the nature and function
of the tumor. Such post translational modifications of the oncofetal
protein are not found in normal cells and do not react with those
monoclonals directed against the original fetal (antigen) protein. The
commercial monoclonals (mAbs) to MUC5ac used to identify cystic
fibrosis products do not recognize the mutated form of the oncofetal
protein and the monoclonals (Neo -101 and 102) developed against
the post translation molecule do not see the cystic fibrosis molecule.
By Immunohistochemistry (IHC), the mAbs that we have developed
for the modified MUC5ac protein only target the tumor protein in
the cancer cell and not in the adjacent normal cell [15,16]. As such
when the mAbs are employed therapeutically, there is a requirement
that the tumor specimen be evaluated by IHC to assure that there is
adequate recognition of the target protein by the monoclonal antibody
being employed for therapy. (Figure 5) illustrated the specific staining
needed to identify the potential of the tumor to respond to treatment.
Figure 4
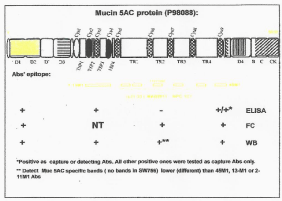
Figure 4
Epitope locations for MUC5ac.
Figure 5
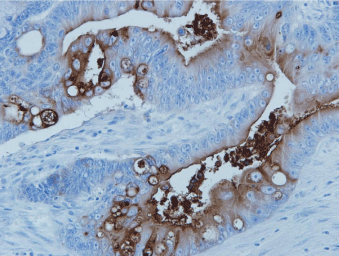
Figure 5
Neo-101-2 staining metastatic colon cancer. Only tumor cells
express the target protein, and when shed, antigen frequently appears in the
bowel lumen where it can be detected by a stool ELISA.
Discussion
Our understanding of the nature of tumor induction took many
years to understand. It was recognized early on, that certain viruses
could induce malignant transformation. This was first noted with the
Rous sarcoma virus transfecting the chicken fibroblast and later with
the Rauscher virus inducing malignancy. It took Varmus, during his
time at NCI, to define the mechanism by which the transformation
process first took place. That is that the virus would enter the DNA
of the nucleus, and in terms of the Rous virus, a defined site on
the chromosome termed the SRC gene would come into play and
function in the mechanism of malignancy [17]. Next it was noted
that when the SRC gene was activated that it induced a neo-protein
that when present would present features of the sarcoma cell that was
nonexistent in the normal fibroblast. This mechanism of induction
was considered to be a prominent feature in most forms of malignant
transformation. Why the tumor continued to progress in patterns of
growth without interruption by the host immune system was again
a matter of speculation. It was not until Prehn had demonstrated in
in-vivo models that the tumor in actuality, behaved as a foreign cell,
not completely different from that of invasion by bacteria and viruses,
that a potential approach toward utilizing the host immune system
for tumor therapy became a reality. The major issue was in defining
the nature of the oncofetal protein that represented the specific
malignancy and making sure that it could be delivered to the host
wherein a proper level of an immune response could be initiated.
Prior to the realization that specific active immunity utilizing
tumor immunogens and their corresponding monoclonals had
the potential to bring many malignancies under control, many
therapeutic products for advanced and recurrent colon cancer were
and still are widely employed. . But despite the development of several
new treatment regimens for this particular form of malignancy
(colorectal cancer) little if any benefit has been appreciated at the
clinical level. A directional change from combination chemotherapy
to immuno-chemotherapy is presently in a transition stage of clinical
development and appears to definitely suggest an improvement in
therapeutic value. . Some immunotherapeutic in 1998, Trastuzumab
(Herceptin) in 1999, Alemtuzumab (Campath) in 2001, Bevacizumab
(Avastin) in 2004, Cetuximab (Erbitux) in 2004, and Panitumumab
(Vectibix) in 2006. More recently, the number of new monoclonal
antibodies on the market has increased with Trametinib, Alectinib,
and Elotuzumab. Many other monoclonal antibodies are currently in
clinical trials as monotherapy.
The ideal monoclonal to employ is the one that can characterize
the tumor via its oncofetal protein. As such the mAb becomes
both diagnostic and therapeutic as seen with the Neo-101 and 102.
Such mabs demonstrating high levels of ADCC can be used alone,
but probably become more effective when used in combination
with other therapies, This monoclonal antibody, when used in the
treatment of solid tumor malignancies should of course target an
immunogenic protein, probably an oncofetal protein characterizing
the tumor rather than a growth factor expressed on the surface of
the tumor but seen as well in many normal tissues. Such oncofetal
proteins do not appear to mutate as we find with the epidermal and
vascular growth factor targets. This latter group can mutate every
4-6 months requiring that the target be reevaluated due to loss of
recognition by the therapeutic antibody Those antibodies used for
targeting an immunogen such as MUC5ac in colon tumors cover a
broad spectrum of transformation processes and therefore can also be
used for defining the premalignant as well as metastatic lesion.
Figure 6
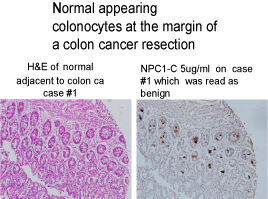
Figure 6
Normal appearing colonocytes that have undergone malignant
transformation and are seen expressing tumor protein.
Summary and Conclusion
The concept of the need for immunotherapy as part of any clinical
study to control advanced colorectal cancer arose from the initial trials
of Hollinshead in the 1980’s. In these studies, pooled allogeneic colon
cancer membrane protein was employed. After 5 years, significant
improvement in survival was noted, but FDA would not allow further
use of such preparations. They realized the possibility that any such
vaccine that could be developed by pooling of tumor specimens
could possibly be contaminated by viruses such as HPV and HIV.
In an attempt to produce a recombinant vaccine, monoclonals were
developed from the original vaccine for immune purification and
sequencing. It became apparent following the development of the
needed monoclonals, that reevaluation of the response to vaccine
data needed to be reviewed.
We quickly recognized at this time, that the expected cell
mediated response in tumor destruction, played only a small role. It
appeared that the major mechanism for tumor destruction was via
ADCC and not via a cytotoxic T cell response As such those mAbs
that we had developed for other needed uses, were now evaluated for
their therapeutic potential.
The first in human investigational study of NPC-1C (Study #
NEO-0901) using NEO-101, was initiated in patients with advanced
colorectal cancer. Pre-clinical testing showed minimal toxicities. In
26 subjects treated in this study, side effects possibly attributable
to NPC-1C (Neo 101) included hives, chills, flushing, shortness of
breath, lower back pain, hematuria, elevated creatinine, elevated LDH,
decreased haptoglobin, and anemia. This was virtually eliminated
when monoclonal Neo-102 was used to replace the original version.
After administration of NPC-1C (NEO-101) to 26 subjects in the
NEO-0901 study, the glycoengineered form of NPC-1C, called NEO-
102, was manufactured for improved stability and decreased RBC
agglutination.
The clinical trial, NEO-0901, dose escalation phase was reinitiated
with the new formulation, and the first combination
clinical trial using NPC-1C and gemcitabine (Study # PB-1201) is
underway.NEO-102 has been administered as monotherapy in 100
patients with colorectal cancer with a very favorable toxicity profile.
The trials employing the high production system mAb Neo 102,
were established and have now completed the Phase II studies and
as reported have shown indications of a significant benefit to the
host [18]. Since the MUC5ac related antigen is the most common
immunogen found among the tumor specimens examined, we are
specifically targeting that protein, a post translational modification of
MUC5ac with its corresponding mAb Neo 102.
The work performed as part of the clinical trial defined the
sequence of the mutated MUC5ac protein and provided the
peptidomimetics of an NPC-1 epitope derived from MUC5ac. This
included composition comprising the amino acid sequence of the
epitope binding site defined by Phage display. That is the polypeptide:
F(PHE) P(PRO) E(GLU) D(ASP) Y(TYR) F(PHE) R(ARG) Y(TYR)
T(THN(ASN) Q((GLN) K(GLY). This sequence proved to be that
fragment of the 600kd MUC5ac molecule responsible for antibody
production and thus could potentially be utilized for eventual peptide
vaccine therapy as a preventative post surgical resection in high risk
patients.
At the 2015 ASCO GI Symposium [19] results of the Phase
1 study of Ensituximab (Neo-102) in chemotherapy refractory
metastatic colorectal Cancer Study were presented in a poster session.
The study revealed a maximal tolerated dose of 3.0 mg/kg IV every 2
weeks. The overall survival observed in this study demonstrated 10.4
months comparing favorably to the historical control for a similar
population of patients with advanced colorectal Ca (5 months). This
led to a larger Phase 2 multi-center colorectal cancer study using
Ensituximab in the same patient population. This study is finalizing
with the results employed in planning for the Phase 3 study. We
are planning to introduce the immunotherapy at the time of initial
recurrence where the immunotherapy will be given in conjunction
with chemotherapy. The latter will be given in doses that do not
suppress the immunocytes but will function to minimize to shedding
of inhibitory molecules produced by the tumor that might interfere
with optimum tumor control.
References
- Stutman O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science.1974; 183: 534–536.
- Hewitt H, Blake B, Walder ASA. critique of the evidence for active host defence against cancer, based on personal studies of 27 murine tumours of spontaneous origin. Br J Cancer. 1976; 33: 241–259.
- Arlen M. Escape Mechanisms employed by tumor cells to allow for growth, invasion and metastasis In Chemoimmunosensitization of Resistant Tumor Cells to Cell Death by Apoptosis. Bonavida. 2006; 243-261.
- Prehn RT, and Main JM. Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst. 1957; 18: 769–778.
- Hollinshead A, Elias G, Arlen M, Mosely MS, Scherrer J. Specific Active Immunotherapy in Patients with Adenocarcinoma of the Colon using TAA – Cancer. 1985; 56: 480-489.
- Elias EG, Hollinshead A, Arlen M, Mosely MS. Adjuvant-Specific Active Immunotherapy in Patients with Colon Adenocarcinoma Utilizing Polypeptide Tumor Associated Antigens (TAA): Proceedings Am Soc Clin Oncol. 1985; 76.
- Hollinshead A, Elias EG, Arlen M, Buda B, Mosley M, Scherrer J. Specific active immunotherapy in patients with adenocarcinoma of the colon utilizing tumor-associated antigens (TAA). A phase I clinical trial. Cancer. 1985; 56: 480-489.
- King DJ. Applications and engineering of monoclonal antibodies. London: CRC Press. 1998.
- Tsang KY, Johnson E, Bishop L, LaVia, Warren RQ, Christian M, et al. Monoclonal Antibodies To Human Colo 16 Carcinoma-Associated Antigens .Abst. Cancer Detection and Prevention. International Soc. Preventive Oncology. 1987; 11.
- Arlen M, Tsang KY. The Nature of the Monoclonal Antibodies Derived from Immunogenic Membrane Antigen of Human Colon Carcinoma Origin. Oncology. 1990; 5: 313-319.
- SP Patel, A Bristol, O Saric. Anti-tumor activity of a novel monoclonal antibody, NPC-1C, optimized for recognition of tumor antigen MUC5AC variant in preclinical models. 2013.
- Stern M, Herrmann R. Overview of monoclonal antibodies in cancer therapy: present and promise. Crit Rev Oncol Hematol. 2005; 54: 11-29.
- Arlen M, Saric O, Wang X, Dubeykonskiy A, Arlen P. Nannocytology vs. Immunohistochemistry of Intestinal Colonocytes to asses the risk of Colon Cancer Based on Field Cancerization. J Cancer. 2013; 4: 165-169.
- Hollinshead A, Arlen M, Arlen P, Bristol A, Luka J, Kantor J, et al. The Dual Functionality of Monoclonal Antibodies Derived from Tumor Associated Antigens. J Surg Oncol. 2010.
- Bara J, Chastre E, Mahiou J, Singh RL, Forgue-Lafitte ME, Hollande E, Godeau F. Gastric M1 mucin, an early oncofetal marker of colon carcinogenesis, is encoded by the MUC5AC gene. Int J Cancer. 1998; 75: 767-773.
- Lau SK, Weiss LM, Chu PG. Differential expression of MUC1, MUC2 and MUC5AC in carcinomas of various sites. Am. J Clin Path. 2004; 122: 61-69.
- Martin GS, Oncogene. 2004; 23: 7910–7917.
- Morse MA, Diaz LA, Azad NS. A Phase 1 study of NPC-1C: a novel, therapeutic antibody to treat pancreas and colorectal cancer. Poster session presented at: American Associate for Cancer Research, Annual Meeting. 2013; 6-10.
- Patel SP, Morse MA, Beatson MA. A Phase I/IIA study of NPC-1C: a novel therapeutic antibody to treat pancreas and colorectal cancer. Poster session presented at: American Society of Clinical Oncology, Annual Meeting. 2013.