Research Article
C6-Ceramide (C6-Cer) to Induce Sensitivity to Cetuximab (Cet) in KRAS Mutant Colorectal Cancer (Crc)
Menendez A1,2, Haidemenos G2, Curzake D3, Luo L2 and Wanebo H1,4*
1Hematology and Medical Oncology, Indiana University School of Medicine, USA
2Department of Medicine, Roger Williams Medical Center, USA
3University of Rhode Island College of Pharmacy, USA
4Division of Surgical Oncology, Landmark Medical Center, USA
*Corresponding author: Harold Wanebo, Hematology and Medical Oncology, Indiana University School of Medicine, Woonsocket, RI, 116 Poppasquash Road, Bristol, Rhode Island, 02809, USA
Published: 26 Jul, 2016
Cite this article as: Menendez A, Haidemenos G, Curzake
D, Luo L, Wanebo H. C6-Ceramide (C6-
Cer) to Induce Sensitivity to Cetuximab
(Cet) in KRAS Mutant Colorectal
Cancer (Crc). Clin Oncol. 2016; 1:
1034.
Abstract
Background: Cet is beneficial for patients with metastatic KRAS Wild type (WT) CRC. C6-Cer
can act synergistically with chemotherapy to induce apoptosis. The aim was to compare growth
inhibition percentage (GIP) of cytostatics 5-FU, oxaliplatin (Ox) and Cet with or without C6-Cer in
KRAS WT and KRAS mutant (KRAS Mut) CRC cell lines (SW48 and SW480, respectively).
Methods: Cells were incubated with IC50 (0.8μM for 5-FU, 0.04μM for Ox, 25 μg/mL for Cet, and
C6-Cer concentrations ranged from 5 to 10 μM). Cell survival was assessed 72h after using 0.4%
Trypan Blue.
Results: C6-Cer’s GIP was 78.3% for SW-480 (vs. 33.33% for SW-48). Addition of C6-Cer increased
GIP with an especially significant effect on SW-480. Addition of 5 and 7.5μM resulted in GIP of
75% and 86.25%, respectively, vs. 32.5% of 5-FU + Ox + Cet alone. The greatest effect was seen
when 10μM of C6-Cer was added (92.5%). Same concentration of drugs increased GIP for SW-48
to 93.5%.
Conclusion: C6-Cer appears to have direct inhibitory properties, especially on KRAS Mut cells.
When added to Ox, 5-FU and Cet, C6-Cer reversed the apparent insensitivity of KRAS Mut to Cet.
Also, the study showed C6-Cer can provide additional synergism to their cytostatic properties in
KRAS WT CRC cell lines. The effect of isolated C6-Cer on KRAS Mut raises possibility of a different
pathway that could bypass EFGR pathway.
Keywords: C6-Cer; Cet; Crc; KRAS
Introduction
CRC is the third most common cancer in the United States and the second leading cause of cancer-related deaths [1]. Metastatic disease is found in 40-50% of patients at the time of diagnosis, with 25% of newly-diagnosed patients found to have liver metastases [2]. Management of patients with (mCRC) is variable, and depends on a number of factors, including performance status and whether the tumor expresses a mutation to KRAS. KRAS mutations have been documented in as high as 35-40% of mCRC patients [3], with a substitution of glycine for aspartate at codon 12 being the most common mutation [4,5]. KRAS mutations cause inhibition of the guanosine-5’- triphosphate (GTP)-ase activity, thereby causing a buildup of GTP-bound KRAS, which is the active form of the protein [5]. Because KRAS is a protein in the pathway between the Epidermal Growth Factor Receptor (EGFR) and oncogenic effects such as cell proliferation and transformation, accumulation of the active form of KRAS allows cells expressing Mut KRAS to promote these effects independent of EGFR activation, which renders drugs that inhibit EGFR much less effective [5]. Due to the fact that surgical resection is rarely a viable option in Stage IV and recurrent colorectal cancer, chemotherapy is the mainstay of treatment for these patients. Combinations of 5-FU with leucovorin, irinotecan or oxaliplatin with or without bevacizumab are all considered standard chemotherapeutic regimens for metastatic disease, and in 2004 the EGFR inhibitor Cetuximab (Cet) received FDA approval after emerging as a promising therapy for patients with metastatic colorectal cancer. Cet is a monoclonal antibody that binds to EGFR to inhibit signal transduction that was approved as first-line treatment in combination with FOLFIRI (leucovorin, 5-FU and irinotecan) for patients with KRAS WT mCRC [6,7]. Subsequent studies however proved Cet to be ineffective on KRAS-Mut mCRC [8]. Ceramide is a sphingolipid metabolite that can induce cancer cell death. It is generated endogenously by ionizing radiation or chemotherapy through the actions of sphingomyelinases. It can also be administered as short-chain analogs, also known as C6-Cer [9]. When exogenous C6-Cer is encapsulated in a nanoliposomal formulation, one in vitro study demonstrated increase in its potency and efficacy [10]. In this study, concentration of C6-Cer required to inhibit 50% of cells (IC50) decreased from 12μmol/L to 5μmol/L when integrated into a nanoliposome [10]. A separate study later found that the mechanism for nanoliposomal C6-Cer-mediated apoptosis was by inhibition of synthesis of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [11]. In vitro analyses have been published regarding the ability of C6- Cer to act synergistically with chemotherapeutic drugs administered for the treatment of several types of cancer [12]. These studies have also shown that C6-Cer may have the potential to inhibit the mutated KRAS ERK/MAPK pathway, and thus reverse the resistance of cancer cells to certain cytotoxic drugs even those that might be EGFR dependent [13]. In another study it was found that C6-Cer inhibited growth of the CRC and induced apoptosis, an effect that was not seen in human mesenchymal stem cells [14]. It is the hypothesis of this experiment that C6-Cer can restore the anti-tumor effect of Cet in patients with the KRAS mutation.
Table 1
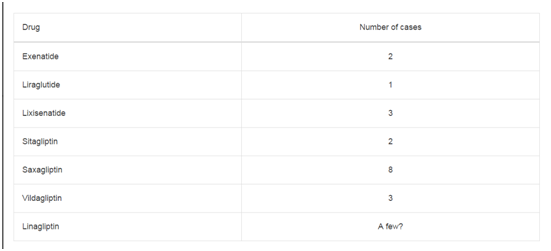
Table 1
Dosing of the combination of oxaliplatin, 5-FU and Cet.
Methodology
Cell cultures
The study is a test system measuring the cell growth of KRAS
WT and KRAS Mut cells. KRAS WT SW-48 (SW48 [SW48] (ATCC®
CCL231TM)) and KRAS-Mut CRC cells SW480 ([SW480] (ATCC®
CCL228TM)) colorectal cancer cell lines (CRC cell lines) were obtained
from American Type Culture Collection (ATCC). They were cultured
in Dulbecco’s modified Eagle’s medium culture (DMEM; Gibco) with
additional sodium bicarbonate (2 grams) (Gibco) and fetal bovine
serum at 10% (Gibco). 100 units/mL Penicillin and 100 microgram/
mL of Streptomycin were added to that mix at 1% concentration.
Cells were cultured at 37oC in a humidified atmosphere including 5%
CO2. Cells were grown in 24-well plates.
Drugs
Oxaliplatin, 5-FU and Cet were added to all plates. C6-Cer was
used at 5mcg/cc. C6- Cer was obtained from Avanti Polar Lipids
Inc, Alabaster, Alabama. Liposomal C6-Cer (5:88:718:0PEG2PE:
DOPC: C6Ceramide) consisted of a phospholipid 1, 2-Dioleoyl-
Sn-glycero-3- phosphocholine (DOPC) and a hydrophilic polymer
polyethylene glycol (PEG) conjugated with another phospholipid
(phophatidylethanolamine). Liposomes were reconstituted from
the lyophilized powder using double distilled water. The drugs were
dissolved in 100% dimethylsulfoxide (DMSO) and then diluted in
the media for experiments. In all the experiments, control cells were
incubated with DMSO alone. The final concentration of DMSO was
maintained at 0.2%.
Determination of chemotherapy dosing
To establish the dosing of the combination of oxaliplatin, 5-FU
and Cet used in the experiment, an analysis of the IC50 was performed.
For this analysis, both SW-48 and SW-480 CRC cells were cultured
in 24-well plates, and then exposed to varying concentrations of each
drug. The well-plates were incubated in varying concentrations of
5-FU and oxaliplatin alone. Using a microscope and Tryptan Blue
stain at 0.4%, researchers counted the number of living cells in each of
the wells 72 hours after incubating the cells in the drug combination.
The percentage of cells killed by the combination was been calculated
and the IC50 was determined. IC50 for this two-drug combination
was 0.8 and 0.04μM, respectively. IC50s were used as a benchmark
for the next phase of the experiment, where Cet was added to the
combinations in order to determine the IC50 of the three-drug
combination. In this experiment, 5-FU and oxaliplatin combinations
used were 0.8 and 0.04μM, 0.4 and 0.02μM and 0.2 and 0.01μM,
Cet concentrations used were 5, 12.5, 25, and 50 μg/mL. After 72
hours, cell counts indicated an IC50 of 0.8μM for 5-FU, 0.04μM for
Oxaliplatin and Erbitux at 12.5μg/mL for SW-48 cells. For SW-480
cells, 0.8μM for 5-FU, 0.04μM for Oxaliplatin and Erbitux at 50μg/
mL failed to improve the total percent of inhibition reached by 0.8μM
of 5-FU and 0.04μM of Oxaliplatin. This difference was expected due
to Cet established lack of efficacy in KRAS-Mut cells. Based on these
results, concentrations of 0.8μM for 5-FU, 0.04μM for Oxaliplatin and
Erbitux at 12.5-50 μg/mL were chosen to be used in the experiment
analyzing the effect of C6-Cer on the IC50 of combination therapy.
C6-Cer was added at concentrations varying from 5 to 10 μM (Table
1).
Figure 1
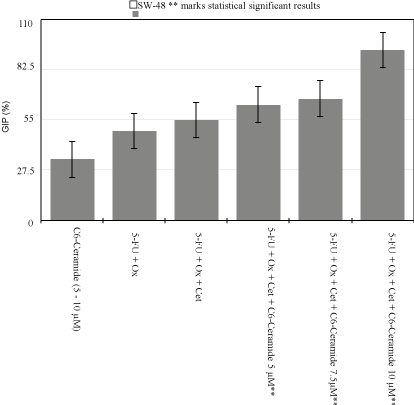
Figure 1
SW-48 Growth Inhibition Percentage Variability.
Results
When compared to DMSO 0.2% control plate, 0.8μM of 5-FU
and 0.04μM of Oxaliplatin alone inhibited 33.5% of SW480 cells
and 49% of SW48 cells from growing. Additionally, 0.8μM of 5- FU,
0.04μM of Oxaliplatin, and Erbitux at 12.5 and 25μg/mL, killed about
28.75%, and 32.5% respectively of KRAS Mut CRC cell lines (SW-
480). Conversely, same concentrations inhibited up to 55% of the
KRAS WT cell line (SW-48) (p<0.005) (Figure 1).
At the lowest C6-Cer concentration of 5μM with IC50 of
chemotherapeutic regimen, cell growth inhibition increased from
32.5% to 75% in KRAS Mut, and from 55% to 63.3% in KRAS WT.
When C6-Cer dose was increased to 7.5μM, the percentage increased
to 86.25% and 66.6% respectively. Finally, when C6-Cer was raised
to 10μM, inhibitory growth percentage reached 92.5% in KRAS Mut,
and 93.3% in KRAS WT (p<0.005) (Figure 2).
C6-Cer was also placed individually with both cell lines
at concentrations ranging from 5 to 10μM. It was noticed the
experimental substance had stronger cytotoxic properties in KRAS
Mut. Inhibitory growth percentage ranged from 70 to 85% in KRAS
Mut cell line, and reached a high of only 40% in KRAS WT.
Discussion
Cet functions as a competitive antagonist to EGFR and may
even lead to its degradation. Direct stimulation of EGFR induces a
conformational change in its extracellular domain that promotes
dimerization with other receptor tyrosine kinases (RTKs), activating
its intrinsic kinase activity and leading to the autophosphorylation of
tyrosine residues, resulting in increased cellular survival, proliferation,
migration and angiogenesis [15]. By inhibiting EGFR, Cet prevents
tumor growth and metastasis and can also induce apoptosis [16].
Cet received accelerated approval after showing it could significantly
reduce the size of tumors and even delay tumor growth for a mean
of 4.1 months when used in combination with irinotecan or even
alone [17]. In the Cet combined with irinotecan in first-line therapy
for mCRC study (CRYSTAL), the efficacy of Cet in combination
with standard FOLFIRI therapy was assessed vs. FOLFIRI alone [8].
This open-label randomized controlled study showed that Cet in
combination with FOLFIRI resulted in statistically significant PFS,
but did not prolong OS [8]. However, in a retrospective analysis of
the study, it was found that Cet produced greater benefits in patients
with WT KRAS. In these patients, OS was increased (23.5 months
vs. 19.5 months in the control group), as well as PFS (9.5 months vs.
8.1 months) and ORR (57% vs. 39%) [8]. This also showed that in
patients with Mut KRAS, Cet did not improve any of these metrics
which led to the indication for Cet in combination with FOLFIRI to
be used as first line therapy in patients with WT KRAS mCRC only.
The CECOG/CORE2 study reported interim results on the efficacy of
Cet in mCRC patients. It was reported that KRAS WT mCRC showed
an increased ORR, OS and PFS with use of a combination of Cet with
FOLFOX4 in comparison to FOLFOX4 alone [18]. This benefit was
not observed in patients with Mut KRAS, lending evidence to the
confirmation that Cet is not effective in patients with Mut KRAS CRC
[18]. And although it can still increase both OS and PFS if mutation is
present, it still remains inferior to the results observed in KRAS WT
patients [19,20]. We witnessed this phenomenon when our results
demonstrated SW-480’s GIP was 33.5% in 5-FU+Ox vs. 32.5% in
5-FU+Ox+Cet at 25μg/mL (Figure 2). Additionally, we replicated the
benefits for KRAS WT CRC, when GIP increased from 49% to 55%
when a small dose of Cet was added to IC50 of drugs (Figure 1). All
of these results were statistically significant and were reproducible in
a second study. A recent pilot study performed at another institution
showed good outcomes of pancreatic cancers, characteristic for
having KRAS gene mutation, exposed to Gemcitabine and Paclitaxel
plus Cet with C6-Cer. Although the effect of C6-Cer was found to
be mainly additive, in vivo studies showed that the combination was
better when it came to stabilization of the tumor volumes and the
survival percentages.
There has been an increase in the study of bioactive lipids as
adjunct components in the field of cancer research. Ceramides are
structural components of the natural cell membrane and they can
induce apoptosis in cancerous cells and even aid in overcoming drug
resistance [21]. It’s most important component to induce cancer
cell death is inhibited by its metabolizing enzyme Glucosylceramide
synthase (GCS) which turns it to a neutral non-apoptotic metabolite
[22]. As a result, several studies have focused on finding ways of
inhibiting GCS, which is hyper-produced in malignant cells [23].
Others have hypothesized even just increasing exogenous C6-Cer
concentrations as a substrate to initiate substantial apoptosis, especially
following affirmations that exogenous C6-Cer administration might
stimulate the generation of intracellular Ceramide [24].
C6-Cer (N-Hexanoyl-D-erythro-sphingosine) possesses
advantageous physical characteristics such as intermediate
hydrophobicity when compared to previously test larger chain
ceramides such as C16-Cer, which are more structurally equivalent
to natural ceramides. In a dose-dependent manner, the analog was
found to activate a cytosolic serine/threonine protein phosphatase,
an intracellular signaling pathway involved in cell differentiation
and proliferation [25]. The nanoliposome triggered intracellular
phosphorylation of PI3K & PKCz and dephosphorylation of PKCa,
resulting in downstream fiber depolymerization, adhesion disassembly
and integrin modulation [26]. Also on par with these effects on cellular
infrastructure, C6-Cer administration has shown direct modulatory
effects on the activation of beta integrins-CD molecules involved
in cell adhesion through phosphorylation inhibition [27]. Others
affirmed ceramides can induce nuclear factor k-B (NF-kB) inhibition,
caspase-3, ADP-ribose polymerase degradation and mitochondrial
cytochrome c release, concluding that apoptosis occurred through
both caspase activation and mitochondrial pathway. It should be
noted, however, that the survival of these cells compared to control
cell lines remained the same, prompting the authors to recognize that
NF-kB inhibition did not modify the ceramide-induced apoptotic
pathway [28]. To add additional evidence of C6-Cer directly affecting
the mitogenic capability of cancer cells, the use and abstinence of serum
growth factors in Molt-4 leukemia cells was studied [29]. They were
able to provide evidence that exogenous C6-Cer was equivalent to the
withdrawal of various serum factors on cell cycle arrest where nearly
80% of cells were arrested in G0/G1, with only 12% leading to apoptotic
cell death. It was proposed that this may be the first preliminary
evidence whereby an intracellular signal transduction pathway
mediated cell cycle arrest, certainly touching on the importance of
lipoid secondary messengers and the possible future target for cancer
therapy. Through another molecular mechanism, C6-Cer has been
shown to have an inhibitory effect on VEGF-induced endothelial cells
[30]. Ceramide administration directly inhibited the endothelial cell
formation and subsequent blood vessel formation. Additionally, it
inhibits both RNA and protein expression of GADPH, an enzyme of
the glycolytic pathway in a CLL model, utilized by cancer cells as part
of their primary metabolism, otherwise known as the ‘Warburg effect’
[31]. This was assessed by concomitantly measuring GADPH and
ATP levels after its administration. They were able to show decreasing
the enzyme levels led to subsequent tumor regression by decreasing
overall protein levels [18]. By inhibiting cancer cell glycolysis, C6-
Cer depletes the cell of adenosine triphosphate (ATP), causing cell
death. The study proved this mechanism by showing that cells that
were pretreated with pyruvate, the end-product of glycolysis, did not
suffer the cytotoxic effects of C6-Cer [18]. This reduction, however,
was not seen in noncancerous peripheral blood mononuclear cells; a
finding that provides evidence for the specificity of C6-Cer to cancer
cells. Overall liposomes are biocompatible and fairly nontoxic to
noncancerous cell lines [32].
With increasing doses of C6-Cer, GIP increased for both cell
lines although not exponentially. It appears SW-480 cells required
less C6-Cer to potentiate the effect of Cet. C6-Cer effect is not only
dose dependent but also cell density and cell type dependent, with an
inverse proportion for the latter ones. Cellular differences between
SW-480 and SW-48 were not studied and could explain the increased
sensitivity of SW-480 cancer cells to C6-Cer, although this would
not subtract importance to the study results. At the highest C6-
Cer concentration of 10μM, GIP rose above 90% in both cell lines,
supporting the theory of C6-Cer reverting the resistance of KRAS Mut
CRC cells to Cet. Furthermore, we also perceived a potentiation of the
Cet synergistic effect with Ox and 5-FU on KRAS WT cells. Initial
analysis showed an increase in GIP from 49 to 55% with addition of
Cet. When C6-Cer was added, GIP increased by 15% at the lowest
dose, and by 69% when high dose C6-Cer was added.
C6-Cer was studied individually. At different concentrations, it
did appear to have certain effect in GIP, especially in KRAS-Mut.
Additional research is warranted to try to establish the molecular
and structural changes in KRAS Mut CRC cells that would explain
for this difference in susceptibility to C6-Cer. Even more, the effect
of isolated C6-Cer in KRAS Mut CRC cell lines raises possibility
of studying a different pathway for treating this type of cancer that
would bypass EGFR receptor pathway. Further studies could include
labeling exogenous C6-Cer within the liposomes to test where is it
that it distributes itself. These results may allow lowering the current
doses of the chemotherapeutics with the combinational therapy
while still maintaining a significant therapeutic effect, resulting in
fewer side effects and even decreased chemoresistance. Additional
studies could include lowering the dose of C6-Cer with the cytostatic
drugs, although the entire panel of C6-Cer adverse effects is yet to be
completely understood.
Administration of C6-Cer as a suspension is unfeasible due to
its hydrophobic nature. Encapsulating the substance in a liposomal
formulation through optimized solvent evaporation technique has
shown decrease cytotoxicity levels when compared to free C6-Cer.
Liposomes are nano particles that enable the delivery of molecules
to particular areas of the organisms through both passive and active
targeting. Adding polyethylene glycol (PEG), a hydrophilic polymer
that enables a shielding effect of those liposomes from the absorption
of proteins, detection by digestive enzymes and the immune system,
leads to longer circulating times, reduced clearance and greater half
lives [33]. Finding a more effective in vivo drug delivering system for
C6-Cer surges as another potential research field.
Figure 2
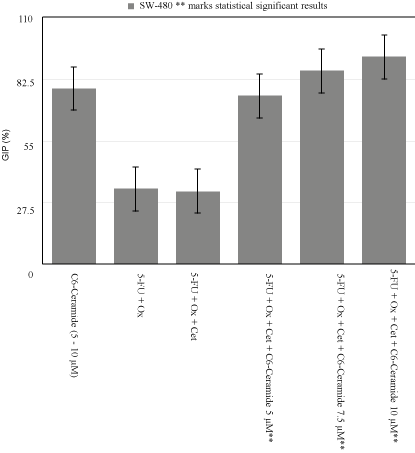
Figure 2
SW-480 Growth Inhibition Percentage Variability.
Figure 3
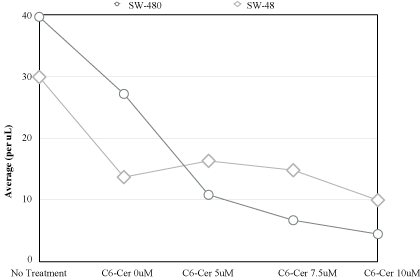
Figure 3
Response comparison to treatment modality.
Conclusion
C6-Cer holds great promise in the future battle against malignancies. From significant effects on intracellular signaling and phosphorylation, to altering enzyme activation and ultimately aiding in programmed cell death, this analog appears to target multiple molecular sites. In our opinion the most important finding of this work is the demonstration that C6-Cer used in combination with 5- FU, oxaliplatin and Cet seems to revert the resistance of KRAS Mut CRC cells lines, although its potentiating effect on KRAS WT CRC cell lines raises a hypothesis of a secondary pathway other that KRAS leading to apoptosis of these cells. We believe the results of this study provide a starting point for clinical studies for C6- Ceramide in patients with relapsing or metastatic KRAS Mut CRC and KRAS WT CRC in combination with standard chemotherapy plus molecular target agents hoping they will translate into clinical benefit for this difficult to treat patient population.
References
- U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999–2011 Incidence and Mortality Web- based Report. Atlanta (GA): Department of Health and Human Services, Centers for Disease Control and Prevention, and National Cancer Institute. 2014.
- Di Fiore F, Sesboue R, Michel P, Sabourin JC, Frebourg T. Molecular determinants of anti-EGFR sensitivity and resistance in metastatic colorectal cacner. Br J Cancer. 2010; 103: 1765-1772.
- Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007; 7: 295-308.
- Guerrero S, Casanova I, Farre L, Mazo A, Capella G, Magues R. K-ras codon 12 mutation induces higher level of resistance of apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene over expression. Cancer Res. 2000; 60: 6750-6756.
- Normanno N, Tejpar S, Morgillo F, De Luca A Van Cutsem E, Giardiello F. Implications of KRAS status and EGFR- targetedtherapies in metastatic CEC. Nat Rev Clin Oncol. 2009; 6: 519-527.
- National Comprehensive Cancer Network. Colon Cancer (Version 2.2015). Accessed 10-7-14.
- National Comprehensive Cancer Network. Rectal Cancer (Version 1.2015). Accessed 10-7-14.
- Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009; 360: 1408-1417.
- Morad SA, Madigan JP, Levin JC, Abdelmageed N, Karimi R, Rosenberg DW, et al. Tamoxifen magnifies therapeutic impact of ceramide in human colorectal cancer cells independent of p53. Biochem Pharmacol 2013; 85: 1057-1065.
- Stover TC, Sharma A, Robertson GP, Kester M. Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin Cancer Res. 2005; 11: 3465-3474.
- Ryland LK, Doshi UA, Shanmugavelandy SS, Fox TE, Aliaga C, Broeg K, et al. C6-Ceramide nanoliposomes taret the Warburg effect in chronic lymphocytic leukemia. PLos One. 2013; 8: e84648.
- Wanebo HJ, Cao C, Lu S, Shrayer D, Wan Y, Bowen W, et al. Abstract B104: Liposomal C6 Ceramide appears to potentiate chemotoxicity of paclitaxel, gemcitabine and cetuximab against aggressive pancreatic cancer. Molecular Cancer Therapeutics. 2013; 12: B104.
- Qiu L, Zhou C, Sun Y, Di W, Scheffler E, Healey S, et al. Paclitaxel and ceramide synergistically induce cell death with transient activation of EGFR and ERK pathway in pancreatic cancer cells. Oncol Rep. 2006; 16: 907-913.
- Huo HZ, Wang B, Qin J, Guo SY, Gu Y. AMP-activated protein kinase (AMPK)/Ulk1-dependent autophagic pathway contributes to C6 ceramide-induced cytotoxic effects in cultured colorectal cancer HT-29 cells. Mol Cell Biochem. 2013; 378: 171-181.
- Citri A, Yarden Y. EGF-ERBB signaling: towards the systems level. Nat Rev Mol Cell Biol. 2006; 7: 505-516.
- Vincenzi B, Zoccoli A, Pantano F, Venditti O, Galluzzo S. Cetuximab: from bench to bedside. Curr Cancer Drug Targets. 2010; 10: 80-95.
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004; 351: 337-345.
- Koza I, Wrba F, Vrbanec DJ, Ocvirk J, Ciuleanu TE, Beslija S, et al. Correlation of KRAS status with clinical outcome in patients (pts) with metastatic colorectal cancer (mCRC) treated first-line with FOLFOX6 + cetuximab (FX+C) or FOLFIRI + cetuximab (FF+C): The CECOG/CORE1.2.001 trial. J Clin Oncol. 2009; 27: 15s.
- De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, et al. Association of KRAS pG13D mutation with outcome in patients with chemotherapy refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010; 304; 1812-1820.
- Tejpar S, Bokemeyer C, Celik I, Schlinchting M, Sartorius U, Van Cutsem E. Influence of KRAS G13D mutations on outcome in patients with metastatic colorectal cancer (mCRC) treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012; 30: 3570-3575.
- Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism—a strategy for overcoming drug resistance. J Nat Cancer Inst. 2001; 93: 347-357.
- Pettus BJ, Chalfant CE, Hannun YA. Ceramide in apoptosis: an overview and current perspectives. Biochimica et Biophysica Acta. 2002; 1585: 114-125.
- Gouazé V, Yu JY, Bleicher RJ, Han TY, Liu YY, Wang H, et al. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol cancer ther. 2004; 3: 633-640.
- Chapman JV, Gouazé-Andersson V, Messner MC, Flowers M, Karimi R, Kester M, et al. Metabolism of short-chain ceramide by human cancer cells—Implications for therapeutic approaches. Biochem pharmacol. 2010; 80: 308-315.
- Dobrowsky RT, Hannun YA. Ceramide stimulates a cytosolic protein phosphatase. J Biol Chem. 1992; 267: 5048-5051.
- Zhang P, Fu C, Hu Y, Dong C, Song Y, Song E. C-6 ceramide nanoliposome suppresses tumor metastasis by eliciting PI3K and PKCz tumor-suppressing activities and regulating integrin affinity modulation. Sci Rep. 2015; 5: 9275.
- Lee YG, Lee J, Cho JY. Cell-permeable ceramides act as novel regulators of U937 cell-cell adhesion mediated by CD29, CD98, and CD147. Immunobiology. 2010; 215: 294-303.
- Fillet M, Bentires-Alj M, Deregowski V, Greimers R, Gielen J, Piette J, et al. Mechanisms involved in exogenous C2 and C6-ceramide-induced cancer cell toxicity. Biochemical Pharmacology. 2003; 65: 1633-1642.
- Jayadev S, Liu B, Bielawska AE, Lee JY, Nazaire F, Pushkareva MYu, et al. Role for ceramide in cell cycle arrest. J Biol Chem. 1995; 270: 2047-2052.
- Bansode RR, Ahmedna M, Svoboda KR, Losso JN. Coupling in vitro and in vivo paradigm reveals a dose dependent inhibition of angiogenesis followed by initiation of autophagy by C6-ceramide. Int J Biol Sci. 2011; 7: 629-644.
- Parnham MJ, Wetzig H. Toxicity screening of liposomes. Chem phys lipids. 1993; 64: 263-274.
- Awasthi VD, Garcia D, Goins BA, Phillips WT. Circulation and biodistribution profiles of long-circulating PEG-liposomes of various sizes in rabbits. Int J pharm. 2003; 253: 121-132.